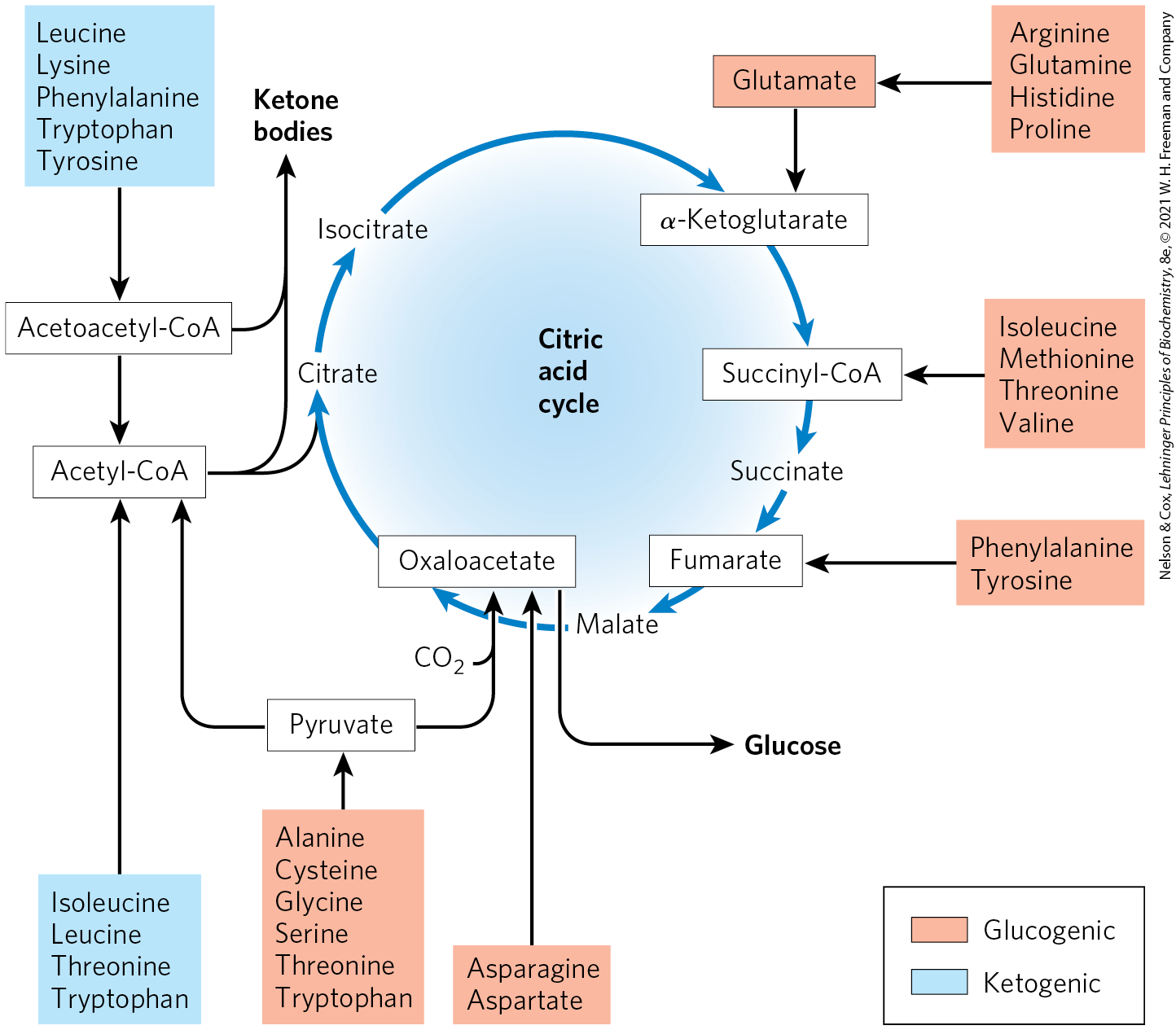
Amino acid catabolism normally accounts for only 10% to 15% of the human body’s energy production; these pathways are not nearly as active as glycolysis and fatty acid oxidation. Flux through these catabolic routes also varies greatly, depending on the balance between requirements for biosynthetic processes and the availability of a particular amino acid. The 20 catabolic pathways converge to form only six major products: pyruvate, acetyl-CoA, α-ketoglutarate, succinyl-CoA, fumarate, and oxaloacetate. All of these enter the citric acid cycle (Fig. 18-15). From here the carbon skeletons are diverted to gluconeogenesis or ketogenesis; alternatively, they are completely oxidized as fuel to and .
FIGURE 18-15 Summary of amino acid catabolism. Amino acids are grouped according to their major degradative end product. Some amino acids are listed more than once because different parts of their carbon skeletons are degraded to different end products. The figure shows the most important catabolic pathways in vertebrates, but there are minor variations among vertebrate species. Threonine, for instance, is degraded by at least two different pathways (see Figs 18-19, 18-27), and the importance of a given pathway can vary with the organism and its metabolic conditions. The glucogenic and ketogenic amino acids are also delineated in the figure, by color shading. Notice that five of the amino acids are both glucogenic and ketogenic. The amino acids degraded to pyruvate are also potentially ketogenic. Only two amino acids, leucine and lysine, are exclusively ketogenic.
We summarize the individual pathways for the 20 amino acids in flow diagrams, each leading to a specific point of entry into the citric acid cycle. In these diagrams, the carbon atoms that enter the citric acid cycle are shown in color. Note that some amino acids appear more than once, reflecting different fates for different parts of their carbon skeletons. Rather than examining every step of every pathway in amino acid catabolism, we single out for special discussion some enzymatic reactions that are particularly noteworthy for their mechanisms or their medical significance.
Some Amino Acids Can Contribute to Gluconeogenesis, Others to Ketone Body Formation
The seven amino acids that are degraded entirely or in part to acetoacetyl-CoA and/or acetyl-CoA — phenylalanine, tyrosine, isoleucine, leucine, tryptophan, threonine, and lysine — can yield ketone bodies in the liver, where acetoacetyl-CoA is converted to acetoacetate and then to acetone and β-hydroxybutyrate (see Fig. 17-16). These are the ketogenic amino acids (Fig. 18-15). Their ability to form ketone bodies is particularly evident in uncontrolled diabetes mellitus, in which the liver produces large amounts of ketone bodies from both fatty acids and the ketogenic amino acids. Ketone bodies may also be metabolized in the brain as fuel in place of glucose in cases during starvation.
The amino acids that are degraded to pyruvate, α-ketoglutarate, succinyl-CoA, fumarate, and/or oxaloacetate can be converted to glucose and glycogen by pathways described in Chapter 14. They are the glucogenic amino acids. The division between ketogenic and glucogenic amino acids is not sharp; five amino acids — tryptophan, phenylalanine, tyrosine, threonine, and isoleucine — are both ketogenic and glucogenic. All amino acids except for lysine and leucine can make some contribution to gluconeogenesis. Catabolism of amino acids is particularly critical to the survival of animals with high-protein diets or during starvation. Leucine is an exclusively ketogenic amino acid that is very common in proteins. Its degradation makes a substantial contribution to ketosis under starvation conditions.
Several Enzyme Cofactors Play Important Roles in Amino Acid Catabolism
The structural diversity of amino acids is reflected in the varied reaction types encountered in their breakdown pathways. We begin our study of these pathways by noting important classes of reactions that recur and introducing their enzyme cofactors. We have already considered one important class: transamination reactions requiring pyridoxal phosphate. One-carbon transfers are another common type of reaction in amino acid catabolism. Such transfers usually involve one of three cofactors: biotin, tetrahydrofolate, or S-adenosylmethionine (Fig. 18-16). These cofactors transfer one-carbon groups in different oxidation states: biotin transfers carbon in its most oxidized state, (see Fig. 14-17); tetrahydrofolate transfers one-carbon groups in intermediate oxidation states and sometimes as methyl groups; and S-adenosylmethionine transfers methyl groups, the most reduced state of carbon. The latter two cofactors are especially important in amino acid and nucleotide metabolism.
FIGURE 18-16 Some enzyme cofactors important in one-carbon transfer reactions. The nitrogen atoms to which one-carbon groups are attached in tetrahydrofolate are shown in blue.
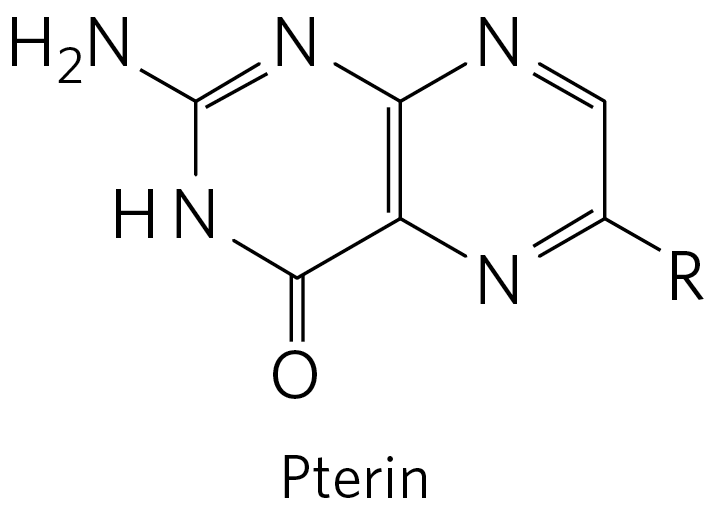
Tetrahydrofolate , synthesized in bacteria, consists of substituted pterin (6-methylpterin), p-aminobenzoate, and glutamate moieties (Fig. 18-16).
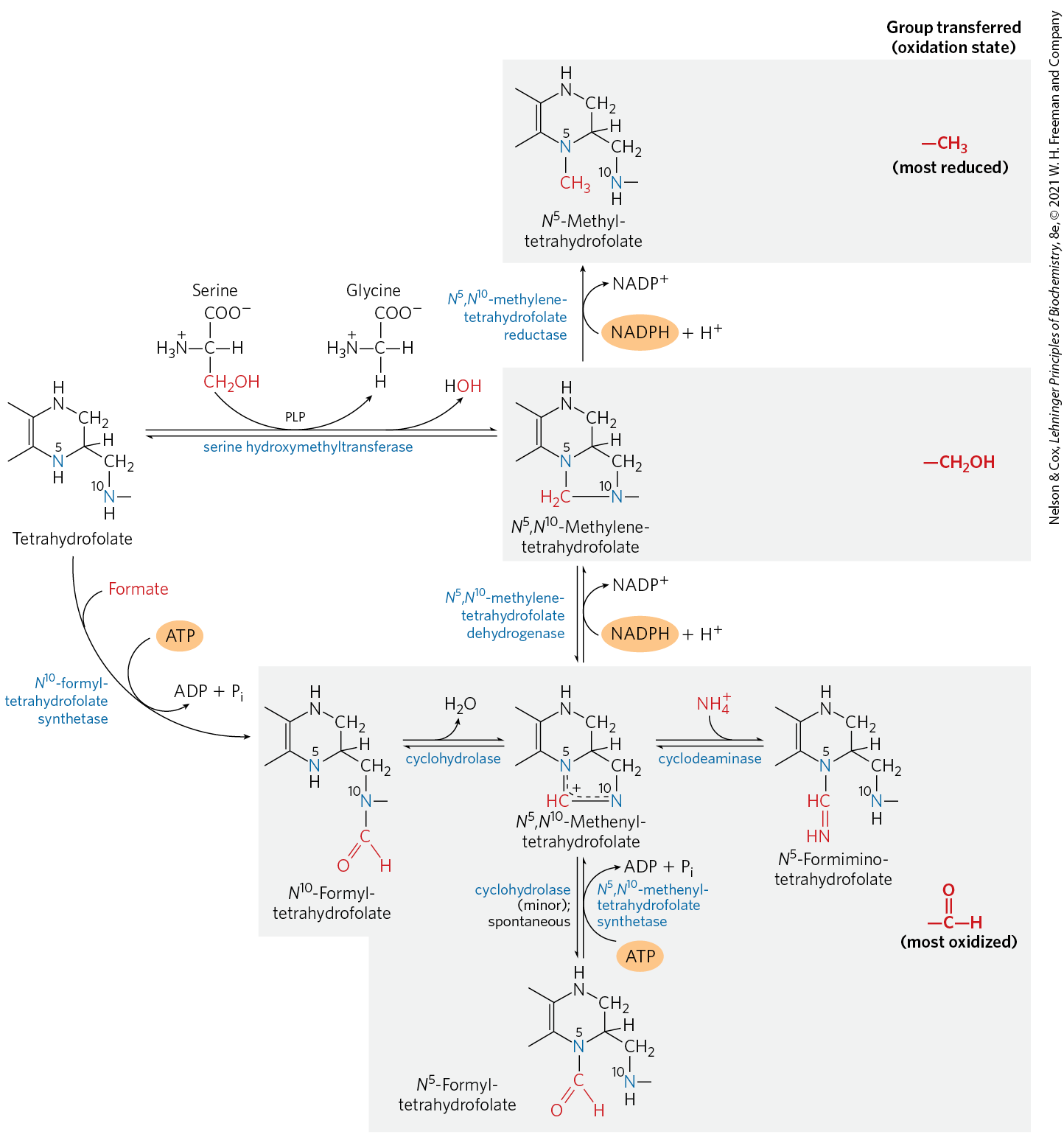
The oxidized form, folate, is a vitamin for mammals; it is converted in two steps to tetrahydrofolate by the enzyme dihydrofolate reductase. The one-carbon group undergoing transfer, in any of three oxidation states, is bonded to N-5 or N-10 or both. The most reduced form of the cofactor carries a methyl group, a more oxidized form carries a methylene group, and the most oxidized forms carry a methenyl, formyl, or formimino group (Fig. 18-17). Most forms of tetrahydrofolate are interconvertible and serve as donors of one-carbon units in a variety of metabolic reactions. The primary source of one-carbon units for tetrahydrofolate is the carbon removed in the conversion of serine to glycine, producing -methylenetetrahydrofolate.
FIGURE 18-17 Conversions of one-carbon units on tetrahydrofolate. The different molecular species are grouped according to oxidation state, with the most reduced at the top and most oxidized at the bottom. All species within a single shaded box are at the same oxidation state. The conversion of -methylenetetrahydrofolate to -methyltetrahydrofolate is effectively irreversible. The enzymatic transfer of formyl groups, as in purine synthesis (see Fig. 22-35) and in the formation of formylmethionine in bacteria (Chapter 27), generally uses -formyltetrahydrofolate rather than -formyltetrahydrofolate. The latter species is significantly more stable and therefore a weaker donor of formyl groups. -Formyltetrahydrofolate is a minor byproduct of the cyclohydrolase reaction, and can also form spontaneously. Conversion of -formyltetrahydrofolate to -methenyltetrahydrofolate requires ATP, because of an otherwise unfavorable equilibrium. Note that -formiminotetrahydrofolate is derived from histidine in a pathway shown in Figure 18-26.
Although tetrahydrofolate can carry a methyl group at N-5, the transfer potential of this methyl group is insufficient for most biosynthetic reactions. S-Adenosylmethionine (adoMet) is the preferred cofactor for biological methyl group transfers. It is synthesized from ATP and methionine by the action of methionine adenosyl transferase (Fig. 18-18, step ). This reaction is unusual in that the nucleophilic sulfur atom of methionine attacks the carbon of the ribose moiety of ATP rather than one of the phosphorus atoms. Triphosphate is released and is cleaved to and on the enzyme, and the is cleaved by inorganic pyrophosphatase; thus three bonds, including two bonds of high-energy phosphate groups, are broken in this reaction. The only other known reaction in which triphosphate is displaced from ATP occurs in the synthesis of coenzyme (see Box 17-2, Fig. 3).
FIGURE 18-18 Synthesis of methionine and S-adenosylmethionine in an activated-methyl cycle. The steps are described in the text. In the methionine synthase reaction (step ), the methyl group is transferred to cobalamin to form methylcobalamin, which is the methyl donor in the formation of methionine. S-Adenosylmethionine, which has a positively charged sulfur (and is thus a sulfonium ion), is a powerful methylating agent in several biosynthetic reactions. The methyl group acceptor (step ) is designated R.
S-Adenosylmethionine is a potent alkylating agent by virtue of its destabilizing sulfonium ion. The methyl group is subject to attack by nucleophiles and is about 1,000 times more reactive than the methyl group of -methyltetrahydrofolate.
Transfer of the methyl group from S-adenosylmethionine to an acceptor yields S-adenosylhomocysteine (Fig. 18-18, step ), which is subsequently broken down to homocysteine and adenosine (step ). Methionine is regenerated by transfer of a methyl group to homocysteine in a reaction catalyzed by methionine synthase (step ), and methionine is reconverted to S-adenosylmethionine to complete an activated-methyl cycle.
One form of methionine synthase common in bacteria uses -methyltetrahydrofolate as a methyl donor. Another form of the enzyme present in some bacteria and mammals uses -methyltetrahydrofolate, but the methyl group is first transferred to cobalamin, derived from coenzyme , to form methylcobalamin as the methyl donor in methionine formation. This reaction and the rearrangement of l-methylmalonyl-CoA to succinyl-CoA (see Box 17-2, Fig. 1a) are the only known coenzyme –dependent reactions in mammals.
The vitamins and folate are closely linked in these pathways. The anemia observed in the rare deficiency disease pernicious anemia (Box 17-2) can be traced to the methionine synthase reaction. As noted above, the methyl group of methylcobalamin is derived from -methyltetrahydrofolate, and this is the only reaction in mammals that uses -methyltetrahydrofolate. The reaction converting the -methylene form to the -methyl form of tetrahydrofolate is irreversible (Fig. 18-17). Thus, if coenzyme is not available for the synthesis of methylcobalamin, metabolic folates become trapped in the -methyl form.
The anemia associated with vitamin deficiency is called megaloblastic anemia. It manifests as a decline in the production of mature erythrocytes (red blood cells) and the appearance in the bone marrow of immature precursor cells, or megaloblasts. Erythrocytes are gradually replaced in the blood by smaller numbers of abnormally large erythrocytes called macrocytes. The defect in erythrocyte development is a direct consequence of the depletion of the -methylenetetrahydrofolate, which is required for synthesis of the thymidine nucleotides needed for DNA synthesis (see Chapter 22). Folate deficiency, in which all forms of tetrahydrofolate are depleted, also produces anemia, for much the same reasons. The anemia symptoms of deficiency can be alleviated by administering either vitamin or folate.
However, it is dangerous to treat pernicious anemia by folate supplementation alone, because the neurological symptoms of deficiency will progress. These symptoms do not arise from the defect in the methionine synthase reaction. Instead, the impaired methylmalonyl-CoA mutase (see Box 17-2 and Fig. 17-12) causes accumulation of unusual, odd-number fatty acids in neuronal membranes. The anemia associated with folate deficiency is thus often treated by administering both folate and vitamin , at least until the metabolic source of the anemia is unambiguously defined. Early diagnosis of deficiency is important because some of its associated neurological conditions may be irreversible.
Folate deficiency also reduces the availability of the -methyltetrahydrofolate required for methionine synthase function. This leads to a rise in homocysteine levels in blood, a condition linked to heart disease, hypertension, and stroke. High levels of homocysteine may be responsible for 10% of all cases of heart disease. The condition is treated with folate supplements.
Tetrahydrobiopterin, another cofactor of amino acid catabolism, is similar to the pterin moiety of tetrahydrofolate, but it is not involved in one-carbon transfers; instead it participates in oxidation reactions. We consider its mode of action when we discuss phenylalanine degradation (see Fig. 18-24).
Six Amino Acids Are Degraded to Pyruvate
The carbon skeletons of six amino acids — alanine, tryptophan, cysteine, serine, glycine, and threonine — are converted in whole or in part to pyruvate. The pyruvate can then be converted to acetyl-CoA and eventually oxidized via the citric acid cycle, or to oxaloacetate and shunted into gluconeogenesis (Fig. 18-19). Alanine yields pyruvate directly on transamination with α-ketoglutarate, and the side chain of tryptophan is cleaved to yield alanine and thus pyruvate. Cysteine is converted to pyruvate in two steps; one removes the sulfur atom, the other is a transamination. Serine is converted to pyruvate by serine dehydratase. Both the β-hydroxyl and the α-amino groups of serine are removed in this single pyridoxal phosphate–dependent reaction (Fig. 18-20a).
FIGURE 18-19 Catabolic pathways for alanine, tryptophan, cysteine, serine, glycine, and threonine. Carbon atoms here and in subsequent figures are color-coded as necessary to trace their fates. The fate of the indole group of tryptophan is shown in Figure 18-21. Details of most of the reactions involving serine and glycine are shown in Figure 18-20. Several pathways for cysteine degradation lead to pyruvate. The pathway for threonine degradation shown here accounts for only about a third of threonine catabolism (for the alternative pathway, see Fig. 18-27).
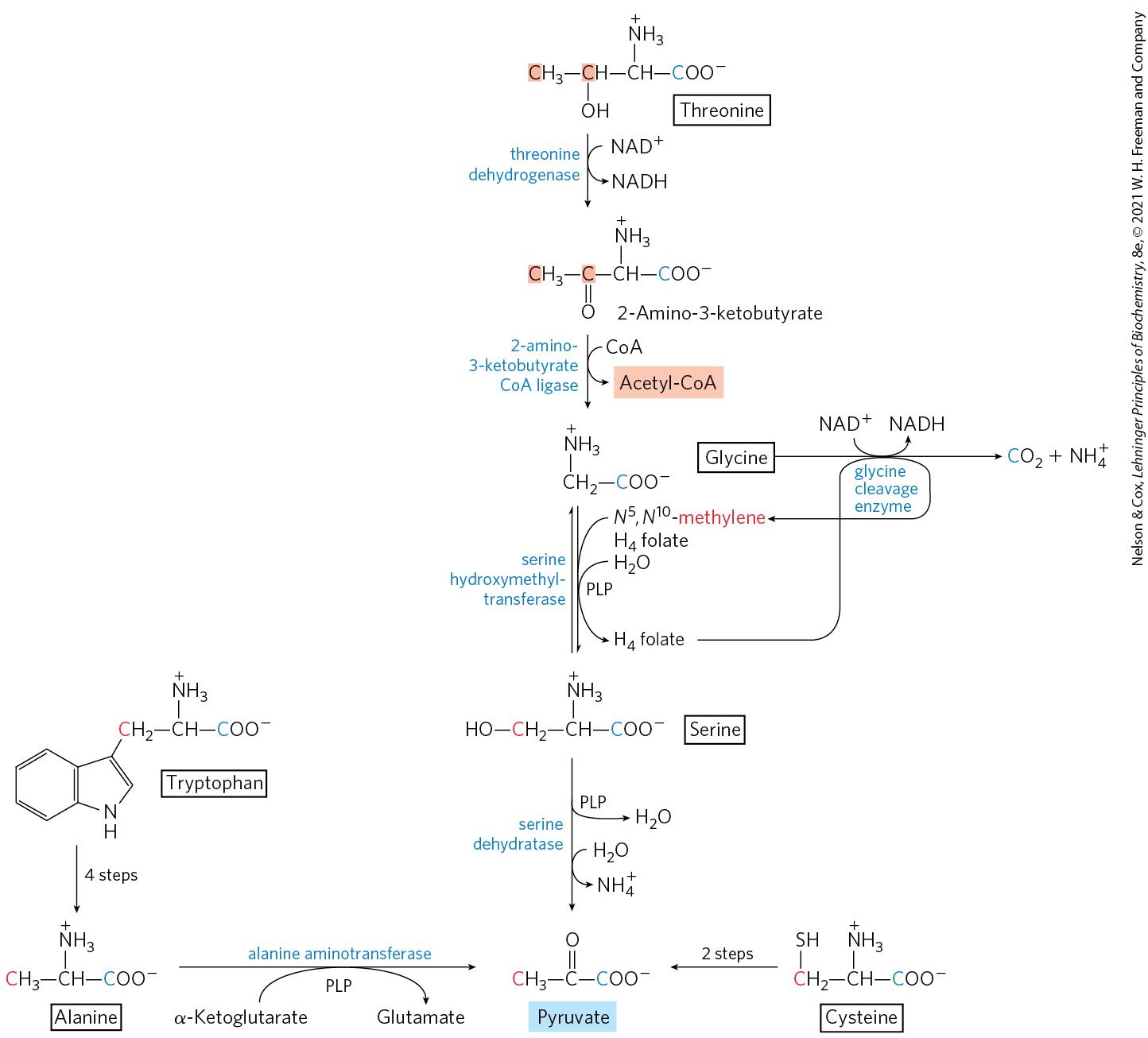
MECHANISM FIGURE 18-20 Interplay of the pyridoxal phosphate and tetrahydrofolate cofactors in serine and glycine metabolism. The first step in each of these reactions (not shown) involves the formation of a covalent imine linkage between enzyme-bound PLP and the substrate amino acid — serine in (a), glycine in (b) and (c). (a) A PLP-catalyzed elimination of water in the serine dehydratase reaction (step ) begins the pathway to pyruvate. (b) In the serine hydroxymethyltransferase reaction, a PLP-stabilized carbanion (product of step ) is a key intermediate in the reversible transfer of the methylene group from -methylenetetrahydrofolate to form serine. (c) The glycine cleavage enzyme is a multienzyme complex, with components P, H, T, and L. The overall reaction, which is reversible, converts glycine to and , with the second glycine carbon taken up by tetrahydrofolate to form -methylenetetrahydrofolate. Pyridoxal phosphate activates the α carbon of amino acids at critical stages in all these reactions, and tetrahydrofolate carries a one-carbon unit in two of them (see Figs 18-6, 18-17).
Glycine is degraded via three pathways, only one of which leads to pyruvate. Glycine is converted to serine by enzymatic addition of a hydroxymethyl group (Fig. 18-19, 18-20b). This reaction, catalyzed by serine hydroxymethyltransferase, requires the coenzymes tetrahydrofolate and pyridoxal phosphate. The serine is converted to pyruvate as described above. In the second pathway, which predominates in animals, glycine undergoes oxidative cleavage to , , and a methylene group (Figs. 18-19, 18-20c). This readily reversible reaction, catalyzed by glycine cleavage enzyme (also called glycine synthase), also requires tetrahydrofolate, which accepts the methylene group. In this oxidative cleavage pathway, the two carbon atoms of glycine do not enter the citric acid cycle. One carbon is lost as and the other becomes the methylene group of -methylenetetrahydrofolate (Fig. 18-17), a one-carbon group donor in certain biosynthetic pathways.
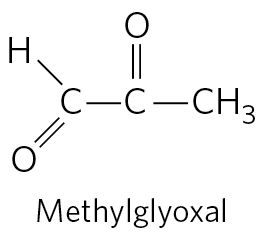
This second pathway for glycine degradation seems to be critical in mammals. Humans with serious defects in glycine cleavage enzyme activity suffer from a condition known as nonketotic hyperglycinemia. The condition is characterized by elevated serum levels of glycine, leading to severe intellectual disability and death in very early childhood. At high levels, glycine is an inhibitory neurotransmitter, which may explain the neurological effects of the disease. Perhaps more important, high levels of glycine increase the levels of 2-amino-3-ketobutyrate, an unstable intermediate in the degradation of threonine in mitochondria (Fig. 18-19). 2-Amino-3-ketobutyrate decarboxylates spontaneously to form the toxic metabolite aminoacetone, which is readily metabolized to the highly reactive methylglyoxal, a molecule that modifies both protein and DNA.
Methylglyoxal is also a byproduct of glycolysis and is implicated in the progression of type 2 diabetes (Box 7-2).
Many genetic defects of amino acid metabolism have been identified in humans (Table 18-2). We shall encounter several more in this chapter.
TABLE 18-2 Some Human Genetic Disorders Affecting Amino Acid Catabolism
Medical condition
Approximate incidence (per 100,000 births)
Defective process
Defective enzyme
Symptoms and effects
Albinism
Melanin synthesis from tyrosine
Tyrosine 3-monooxygenase (tyrosinase)
Lack of pigmentation; white hair, pink skin
Alkaptonuria
Tyrosine degradation
Homogentisate 1,2-dioxygenase
Dark pigment in urine; late-developing arthritis
Argininemia
Urea synthesis
Arginase
Intellectual disability
Argininosuccinic acidemia
Urea synthesis
Argininosuccinase
Vomiting; convulsions
Carbamoyl phosphate synthetase I deficiency
Urea synthesis
Carbamoyl phosphate synthetase I
Lethargy; convulsions; early death
Homocystinuria
Methionine degradation
Cystathionine β-synthase
Faulty bone development; intellectual disability
Maple syrup urine disease (branched-chain ketoaciduria)
Isoleucine, leucine, and valine degradation
Branched-chain α-keto acid dehydrogenase complex
Vomiting; convulsions; intellectual disability; early death
Methylmalonic acidemia
Conversion of propionyl-CoA to succinyl-CoA
Methylmalonyl-CoA mutase
Vomiting; convulsions; intellectual disability; early death
Phenylketonuria
Conversion of phenylalanine to tyrosine
Phenylalanine hydroxylase
Neonatal vomiting; intellectual disability
In the third and final pathway of glycine degradation, the achiral glycine molecule is a substrate for the enzyme d-amino acid oxidase. The glycine is converted to glyoxylate, an alternative substrate for lactate dehydrogenase (p. 526). Glyoxylate is oxidized in an -dependent reaction to oxalate:
The function of d-amino acid oxidase, present at high levels in the kidney, is thought to be the detoxification of ingested d-amino acids derived from bacterial cell walls and from grilled foodstuffs (high heat causes some spontaneous racemization of the l-amino acids in proteins). Oxalate, whether obtained in foods or produced enzymatically in the kidneys, has medical significance. Crystals of calcium oxalate account for up to 75% of all kidney stones.
There are two significant pathways for threonine degradation. One pathway leads to pyruvate via glycine (Fig. 18-19). The conversion to glycine occurs in two steps, with threonine first converted to 2-amino-3-ketobutyrate by the action of threonine dehydrogenase. This pathway is important in a few classes of rapidly dividing human cells, such as embryonic stem cells. The glycine generated by this pathway is broken down primarily by the glycine cleavage enzyme (Fig. 18-19). The -methylenetetrahydrofolate thus generated (Fig. 18-20c) is needed for the synthesis, via pathways described in Chapter 22, of nucleotides used in DNA replication. However, in most human tissues, the degradation of threonine via glycine is a relatively minor pathway, accounting for 10% to 30% of threonine catabolism. It is more important in some other mammals. The major pathway in most human tissues leads to succinyl-CoA, as described later in this chapter.
Seven Amino Acids Are Degraded to Acetyl-CoA
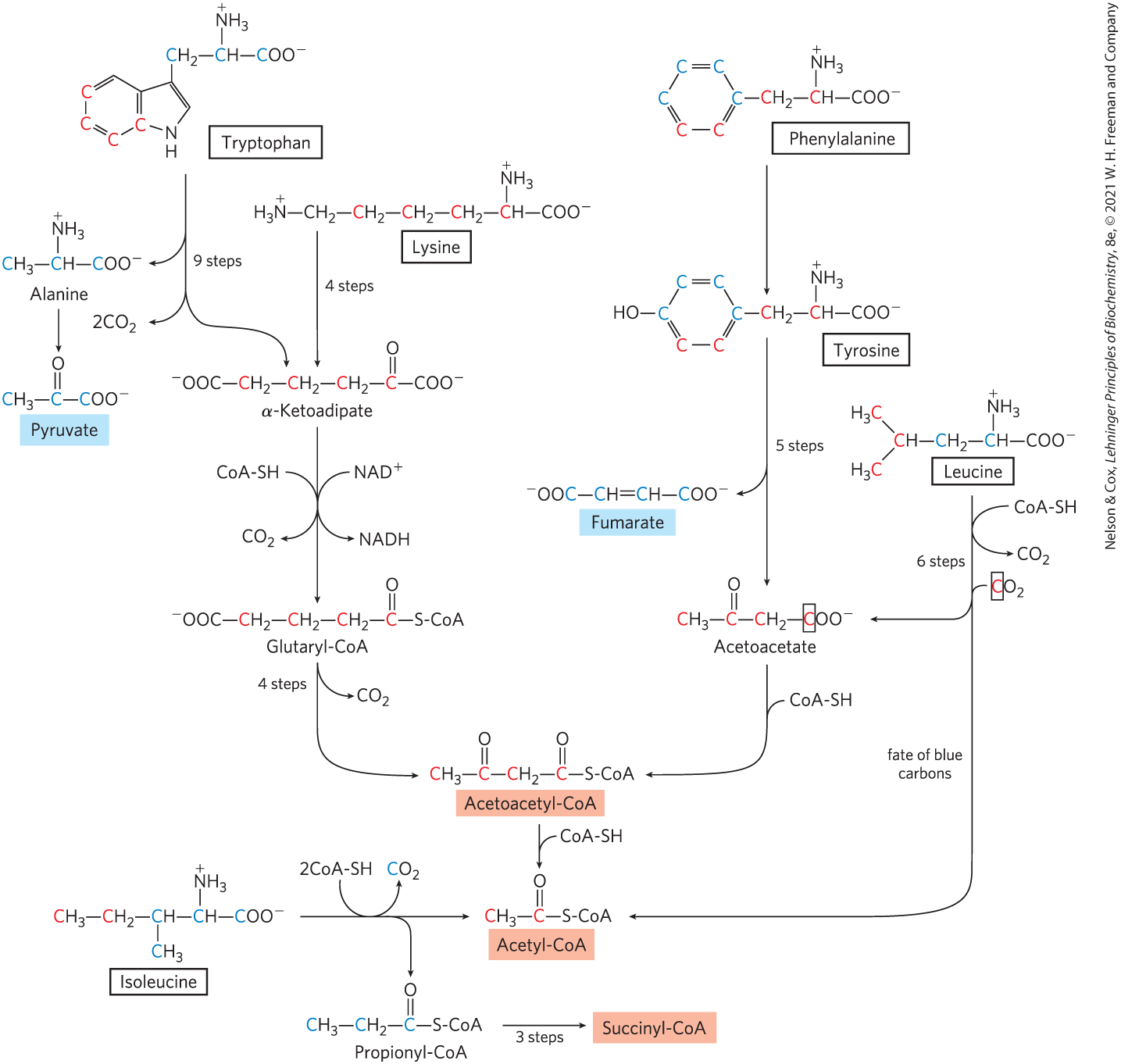
Portions of the carbon skeletons of seven amino acids — tryptophan, lysine, phenylalanine, tyrosine, leucine, isoleucine, and threonine — yield acetyl-CoA and/or acetoacetyl-CoA, the latter being converted to acetyl-CoA (Fig. 18-21). Some of the final steps in the degradative pathways for leucine, lysine, and tryptophan resemble steps in the oxidation of fatty acids (see Fig. 17-9). Threonine (not shown in Fig. 18-21) yields some acetyl-CoA via the minor pathway illustrated in Figure 18-19.
FIGURE 18-21 Catabolic pathways for tryptophan, lysine, phenylalanine, tyrosine, leucine, and isoleucine. These amino acids donate some of their carbons (red) to acetoacetate and acetyl-CoA. In the pathway for leucine catabolism, the carbon in acetoacetate that is donated by is surrounded by a box to distinguish it from the three carbons that come from leucine itself. Tryptophan, phenylalanine, tyrosine, and isoleucine also contribute carbons (blue) to pyruvate or citric acid cycle intermediates. The phenylalanine pathway is described in more detail in Figure 18-23. The fate of nitrogen atoms is not traced in this scheme; in most cases they are transferred to α-ketoglutarate to form glutamate.
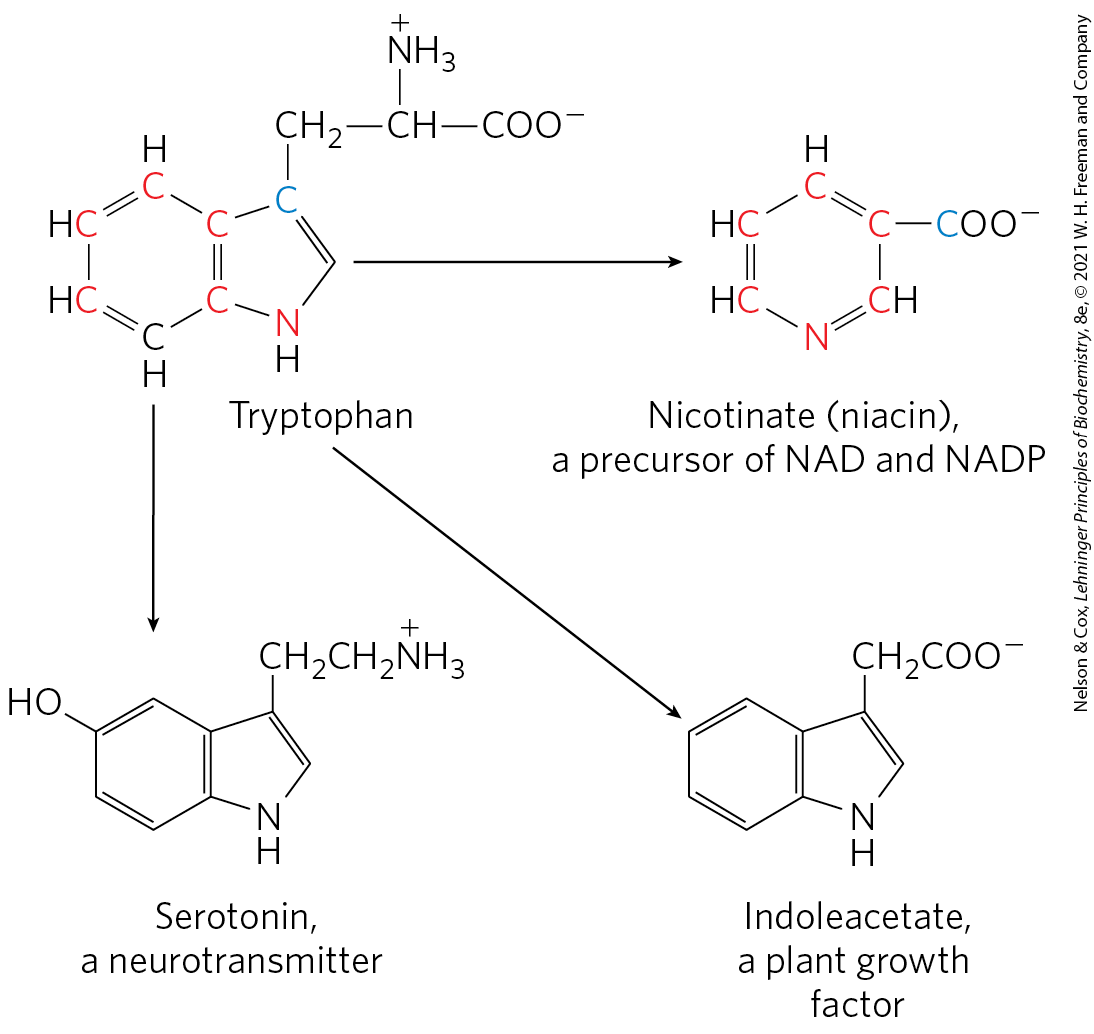
The degradative pathways of two of these seven amino acids deserve special mention. Tryptophan breakdown is the most complex of all the pathways of amino acid catabolism in animal tissues; portions of tryptophan (four of its carbons) yield acetyl-CoA via acetoacetyl-CoA. Some of the intermediates in tryptophan catabolism are precursors for the synthesis of other biomolecules (Fig. 18-22), including nicotinate, a precursor of NAD and NADP in animals; serotonin, a neurotransmitter in vertebrates; and indoleacetate, a growth factor in plants. Some of these biosynthetic pathways are described in more detail in Chapter 22 (see Figs. 22-30, 22-31).
FIGURE 18-22 Tryptophan as precursor. The aromatic rings of tryptophan give rise to nicotinate (niacin), indoleacetate, and serotonin. Colored atoms trace the source of the ring atoms in nicotinate.
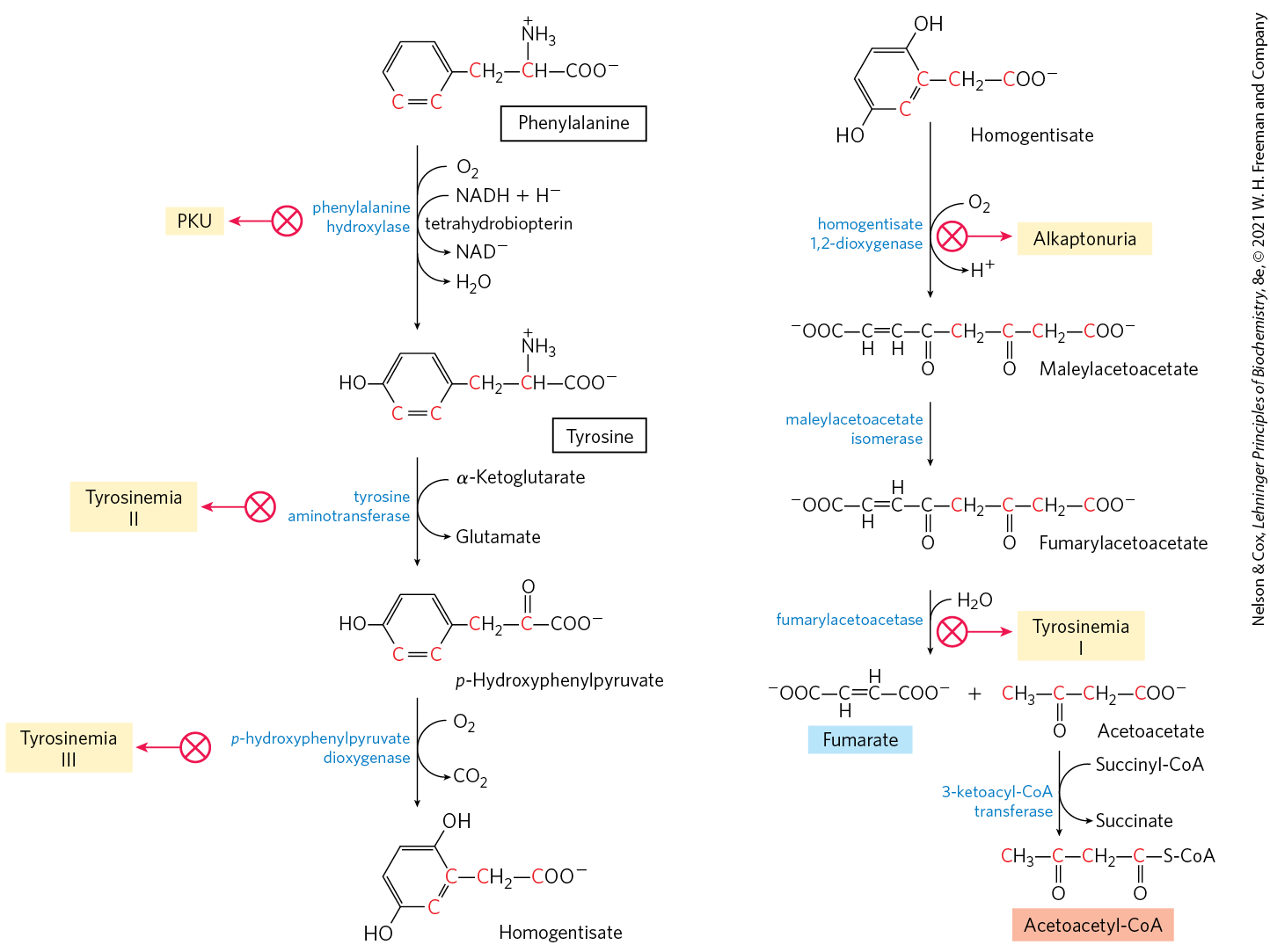
The breakdown of phenylalanine is noteworthy because genetic defects in the enzymes of this pathway lead to several inheritable human diseases (Fig. 18-23), as discussed below. Phenylalanine and its oxidation product tyrosine (both with nine carbons) are degraded into two fragments, both of which can enter the citric acid cycle: four of the nine carbon atoms yield free acetoacetate, which is converted to acetoacetyl-CoA and thus acetyl-CoA, and a second four-carbon fragment is recovered as fumarate. Eight of the nine carbons of these two amino acids thus enter the citric acid cycle; the remaining carbon is lost as . Phenylalanine, after its hydroxylation to tyrosine, is also the precursor of dopamine, a neurotransmitter, and of norepinephrine and epinephrine, hormones secreted by the adrenal medulla (see Fig. 22-31). Melanin, the black pigment of skin and hair, is also derived from tyrosine.
FIGURE 18-23 Catabolic pathways for phenylalanine and tyrosine. In humans these amino acids are normally converted to acetoacetyl-CoA and fumarate. Genetic defects in many of these enzymes cause inheritable human diseases (shaded yellow).
Phenylalanine Catabolism Is Genetically Defective in Some People
Given that many amino acids are either neurotransmitters or precursors or antagonists of neurotransmitters, it is not surprising that genetic defects of amino acid metabolism can cause defective neural development and intellectual deficits. In most such diseases, specific intermediates accumulate. For example, a genetic defect in phenylalanine hydroxylase, the first enzyme in the catabolic pathway for phenylalanine (Fig. 18-23), is responsible for the disease phenylketonuria (PKU), the most common cause of elevated levels of phenylalanine in the blood (hyperphenylalaninemia).
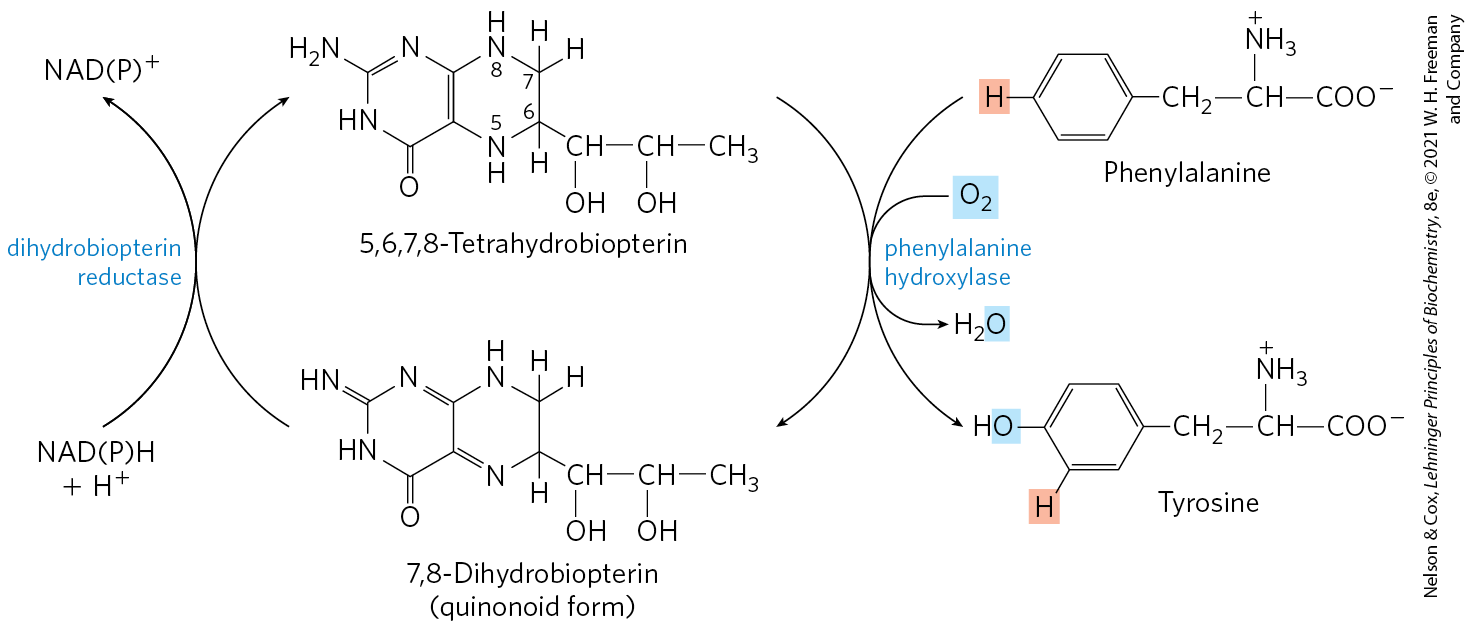
Phenylalanine hydroxylase (also called phenylalanine-4-monooxygenase) is one of a general class of enzymes called mixed-function oxygenases (see Box 21-1), all of which catalyze simultaneous hydroxylation of a substrate by an oxygen atom of and reduction of the other oxygen atom to . Phenylalanine hydroxylase requires the cofactor tetrahydrobiopterin, which carries electrons from NADPH to and becomes oxidized to dihydrobiopterin in the process (Fig. 18-24). It is subsequently reduced by the enzyme dihydrobiopterin reductase in a reaction that requires NADPH.
FIGURE 18-24 Role of tetrahydrobiopterin in the phenylalanine hydroxylase reaction. The H atom shaded pink is transferred directly from C-4 to C-3 in the reaction. This feature, discovered at the National Institutes of Health, is called the NIH shift.
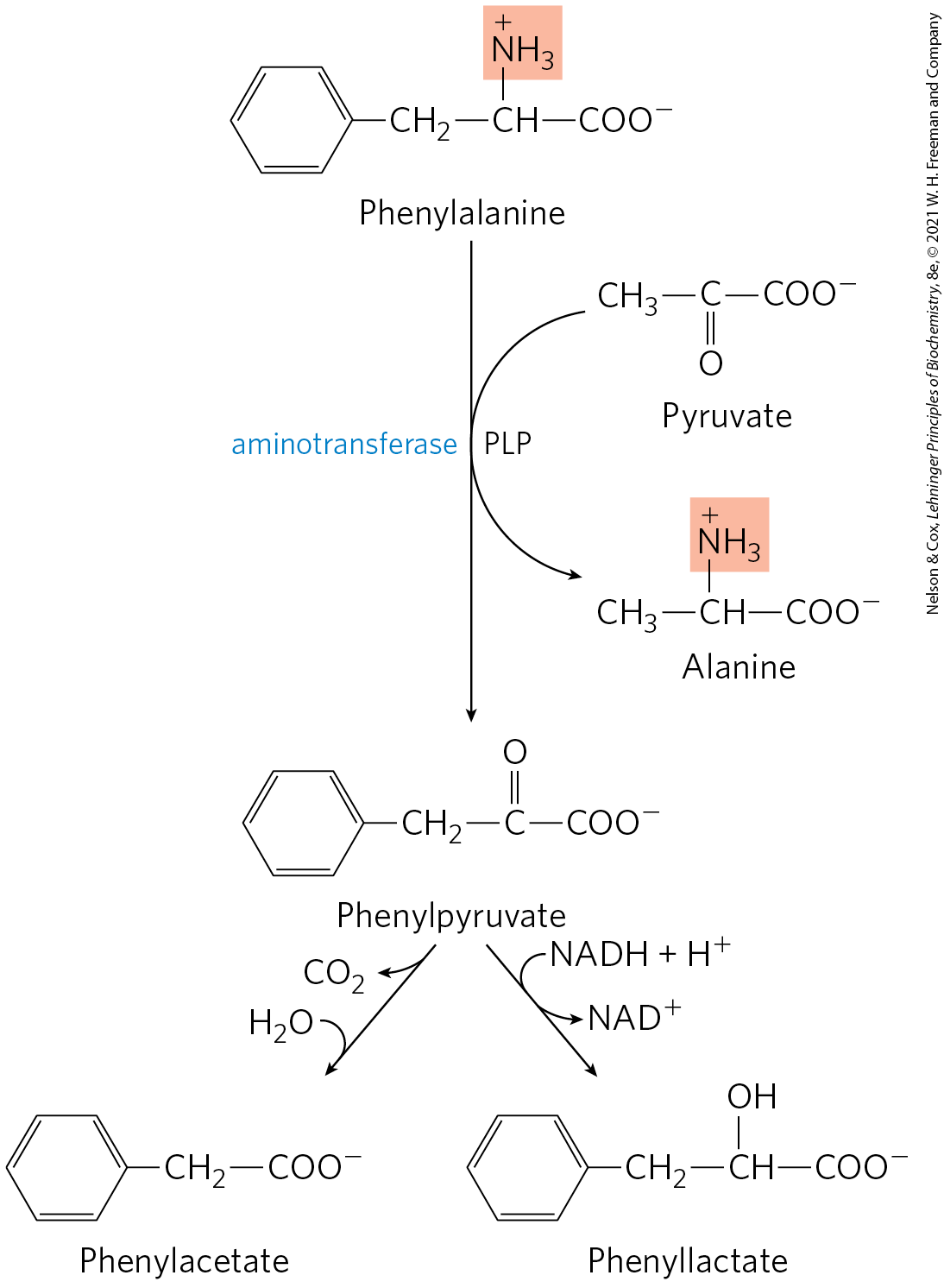
In individuals with PKU, a secondary, normally little-used pathway of phenylalanine metabolism comes into play. In this pathway phenylalanine undergoes transamination with pyruvate to yield phenylpyruvate (Fig. 18-25). Phenylalanine and phenylpyruvate accumulate in the blood and tissues and are excreted in the urine — hence the name “phenylketonuria.” Much of the phenylpyruvate, rather than being excreted as such, is either decarboxylated to phenylacetate or reduced to phenyllactate. Phenylacetate imparts a characteristic honeylike odor to the urine, which nurses have traditionally used to detect PKU in infants. The accumulation of phenylalanine or its metabolites in early life impairs normal development of the brain, causing severe intellectual deficits. This may be caused by excess phenylalanine competing with other amino acids for transport across the blood-brain barrier, resulting in a deficit of required metabolites.
FIGURE 18-25 Alternative pathways for catabolism of phenylalanine in phenylketonuria. In PKU, phenylpyruvate accumulates in the tissues, blood, and urine. The urine may also contain phenylacetate and phenyllactate.
Phenylketonuria was among the first inheritable metabolic defects discovered in humans. When this condition is recognized early in infancy, intellectual disability can be prevented by rigid dietary control. The diet must supply only enough phenylalanine and tyrosine to meet the needs for protein synthesis. Consumption of protein-rich foods must be curtailed. Natural proteins, such as casein of milk, must first be hydrolyzed and much of the phenylalanine removed to provide an appropriate diet, at least through childhood. Because the artificial sweetener aspartame is a dipeptide of aspartate and the methyl ester of phenylalanine (see Fig. 1-23a), foods sweetened with aspartame bear warnings addressed to individuals on phenylalanine-controlled diets.
The dietary restrictions are difficult to follow perfectly for a lifetime, and thus often do not completely eliminate neurological symptoms. An enzyme substitution treatment has been developed in which the enzyme phenylalanine ammonia lyase is modified with polyethylene glycol (PEGylated) and injected subcutaneously to degrade phenylalanine in proteins ingested as part of a somewhat less restricted diet. Derived from plants, bacteria, and many yeast and fungi, phenylalanine ammonia lyase normally contributes to the biosynthesis of polyphenol compounds such as flavonoids. It degrades phenylalanine to the harmless metabolite trans-cinnamic acid and ammonia; the small amounts of ammonia generated are not toxic. The treatment was approved for patients in 2018. The long-term effects of the treatment continue to be studied.
Phenylketonuria can also be caused by a defect in the enzyme that catalyzes the regeneration of tetrahydrobiopterin (Fig. 18-24). The treatment in this case is more complex than restricting the intake of phenylalanine and tyrosine. Tetrahydrobiopterin is also required for the formation of l-3,4-dihydroxyphenylalanine (l-dopa) and 5-hydroxytryptophan — precursors of the neurotransmitters norepinephrine and serotonin, respectively. In phenylketonuria of this type, these precursors must be supplied in the diet, along with tetrahydrobiopterin.
Screening newborns for genetic diseases can be highly cost-effective, especially in the case of PKU. The tests (no longer relying on urine odor) are relatively inexpensive, and the detection and early treatment of PKU in infants (eight to ten cases per 100,000 newborns) saves millions of dollars in later health care costs each year. More importantly, the emotional trauma avoided by early detection with these simple tests is inestimable.
Another inheritable disease of phenylalanine catabolism is alkaptonuria, in which the defective enzyme is homogentisate dioxygenase (Fig. 18-23). Less serious than PKU, this condition produces few ill effects, although large amounts of homogentisate are excreted and its oxidation turns the urine black. Individuals with alkaptonuria are also prone to develop a form of arthritis. Alkaptonuria is of considerable historical interest. Archibald Garrod discovered in the early 1900s that this condition is inherited, and he traced the cause to the absence of a single enzyme. Garrod was the first to make a connection between an inheritable trait and an enzyme — a great advance on the path that ultimately led to our current understanding of genes and the information pathways described in Part III.
Five Amino Acids Are Converted to α-Ketoglutarate
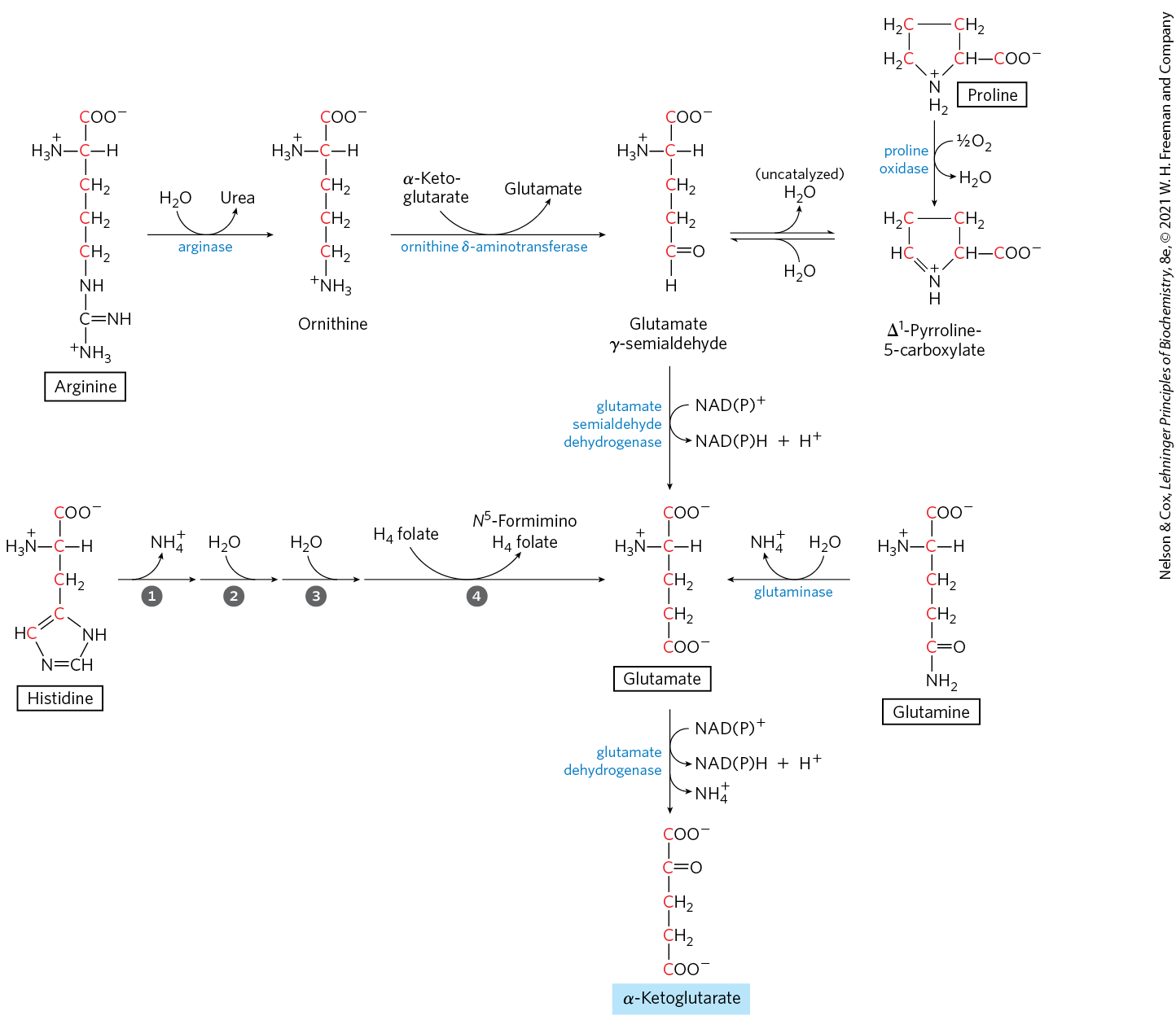
The carbon skeletons of five amino acids (proline, glutamate, glutamine, arginine, and histidine) enter the citric acid cycle as α-ketoglutarate (Fig. 18-26). Proline, glutamate, and glutamine have five-carbon skeletons. The cyclic structure of proline is opened by oxidation of the carbon most distant from the carboxyl group to create a Schiff base, then hydrolysis of the Schiff base to form a linear semialdehyde, glutamate γ-semialdehyde. This intermediate is further oxidized at the same carbon to produce glutamate. The action of glutaminase, or any of several enzyme reactions in which glutamine donates its amide nitrogen to an acceptor, converts glutamine to glutamate. Transamination or deamination of glutamate produces α-ketoglutarate.
FIGURE 18-26 Catabolic pathways for arginine, histidine, glutamate, glutamine, and proline. These amino acids are converted to α-ketoglutarate. The numbered steps in the histidine pathway are catalyzed by histidine ammonia lyase, urocanate hydratase, imidazolonepropionase, and glutamate formimino transferase.
Arginine and histidine contain five adjacent carbons and a sixth carbon attached through a nitrogen atom. The catabolic conversion of these amino acids to glutamate is therefore slightly more complex than the path from proline or glutamine (Fig. 18-26). Arginine is converted to the five-carbon skeleton of ornithine in the urea cycle (Fig. 18-10), and the ornithine is transaminated to glutamate γ-semialdehyde. Conversion of histidine to the five-carbon glutamate occurs in a multistep pathway; the extra carbon is removed in a step that uses tetrahydrofolate as cofactor.
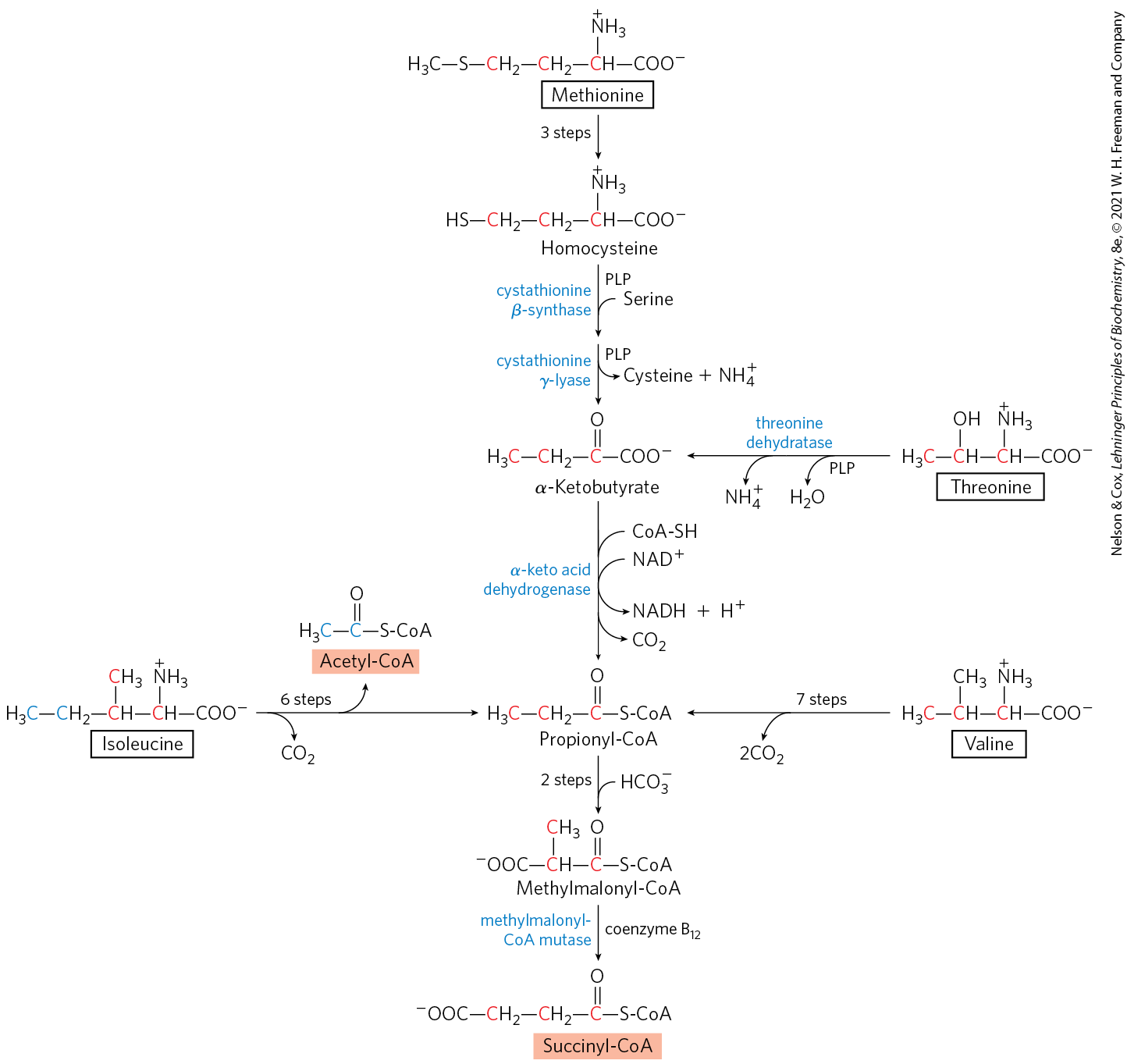
Four Amino Acids Are Converted to Succinyl-CoA
The carbon skeletons of methionine, isoleucine, threonine, and valine are degraded by pathways that yield succinyl-CoA (Fig. 18-27), an intermediate of the citric acid cycle. Methionine donates its methyl group to one of several possible acceptors through S-adenosyl methionine, and three of its four remaining carbon atoms are converted to the propionate of propionyl-CoA, a precursor of succinyl-CoA. Isoleucine undergoes transamination, followed by oxidative decarboxylation of the resulting α-keto acid. The remaining five-carbon skeleton is further oxidized to acetyl-CoA and propionyl-CoA. Valine undergoes transamination and decarboxylation, then a series of oxidation reactions that convert the remaining four carbons to propionyl-CoA. Some parts of the valine and isoleucine degradative pathways closely parallel steps in fatty acid degradation (see Fig. 17-9). In human tissues, threonine is also converted in two steps to propionyl-CoA. This is the primary pathway for threonine degradation in humans (see Fig. 18-19 for the alternative pathway). The mechanism of the first step is analogous to that catalyzed by serine dehydratase, and the serine and threonine dehydratases may actually be the same enzyme.
FIGURE 18-27 Catabolic pathways for methionine, isoleucine, threonine, and valine. These amino acids are converted to succinyl-CoA; isoleucine also contributes two of its carbon atoms to acetyl-CoA (see Fig. 18-21). The pathway of threonine degradation shown here occurs in humans; a pathway found in other organisms is shown in Figure 18-19. The route from methionine to homocysteine is described in more detail in Figure 18-18; the conversion of homocysteine to α-ketobutyrate, in Figure 22-16; and the conversion of propionyl-CoA to succinyl-CoA, in Figure 17-12.
The propionyl-CoA derived from these three amino acids is converted to succinyl-CoA by a pathway described in Chapter 17: carboxylation to methylmalonyl-CoA, epimerization of the methylmalonyl-CoA, and conversion to succinyl-CoA by the coenzyme –dependent methylmalonyl-CoA mutase (see Fig. 17-12). In the rare genetic disease known as methylmalonic acidemia, methylmalonyl-CoA mutase is lacking — with serious metabolic consequences (Table 18-2; Box 18-2).
Branched-Chain Amino Acids Are Not Degraded in the Liver
Although much of the catabolism of amino acids takes place in the liver, the three amino acids with branched side chains (leucine, isoleucine, and valine) are oxidized as fuels primarily in muscle, adipose, kidney, and brain tissue. These extrahepatic tissues contain an aminotransferase, absent in liver, that acts on all three branched-chain amino acids to produce the corresponding α-keto acids (Fig. 18-28). The branched-chain α-keto acid dehydrogenase complex then catalyzes oxidative decarboxylation of all three α-keto acids, in each case releasing the carboxyl group as and producing the acyl-CoA derivative. This reaction is formally analogous to two other oxidative decarboxylations encountered in Chapter 16: oxidation of pyruvate to acetyl-CoA by the pyruvate dehydrogenase complex and oxidation of α-ketoglutarate to succinyl-CoA by the α-ketoglutarate dehydrogenase complex (see Fig. 16-12). In fact, all three enzyme complexes are similar in structure and share essentially the same reaction mechanism. Five cofactors (thiamine pyrophosphate, FAD, NAD, lipoate, and coenzyme A) participate, and the three proteins in each complex catalyze homologous reactions. This is clearly a case in which enzymatic machinery that evolved to catalyze one reaction was “borrowed” by gene duplication and further evolved to catalyze similar reactions in other pathways.
FIGURE 18-28 Catabolic pathways for the three branched-chain amino acids: valine, isoleucine, and leucine. All three pathways occur in extrahepatic tissues and share the first two enzymes, as shown here. The branched-chain α-keto acid dehydrogenase complex is analogous to the pyruvate and α-ketoglutarate dehydrogenase complexes and requires the same five cofactors (some not shown here). This enzyme is defective in people with maple syrup urine disease.
Experiments with rats have shown that the branched-chain α-keto acid dehydrogenase complex is regulated by covalent modification in response to the content of branched-chain amino acids in the diet. With little or no excess dietary intake of branched-chain amino acids, the enzyme complex is phosphorylated by a protein kinase and thereby inactivated. Addition of excess branched-chain amino acids to the diet results in dephosphorylation and consequent activation of the enzyme. Recall that the pyruvate dehydrogenase complex is subject to similar regulation by phosphorylation and dephosphorylation (see Fig. 16-19).
There is a relatively rare genetic disease in which the three branched-chain α-keto acids (as well as their precursor amino acids, especially leucine) accumulate in the blood and “spill over” into the urine. This condition, called maple syrup urine disease because of the characteristic odor imparted to the urine by the α-keto acids, results from a defective branched-chain α-keto acid dehydrogenase complex. Untreated, the disease results in abnormal development of the brain and death in early infancy. Treatment entails rigid control of the diet, limiting the intake of valine, isoleucine, and leucine to the minimum required to permit normal growth.
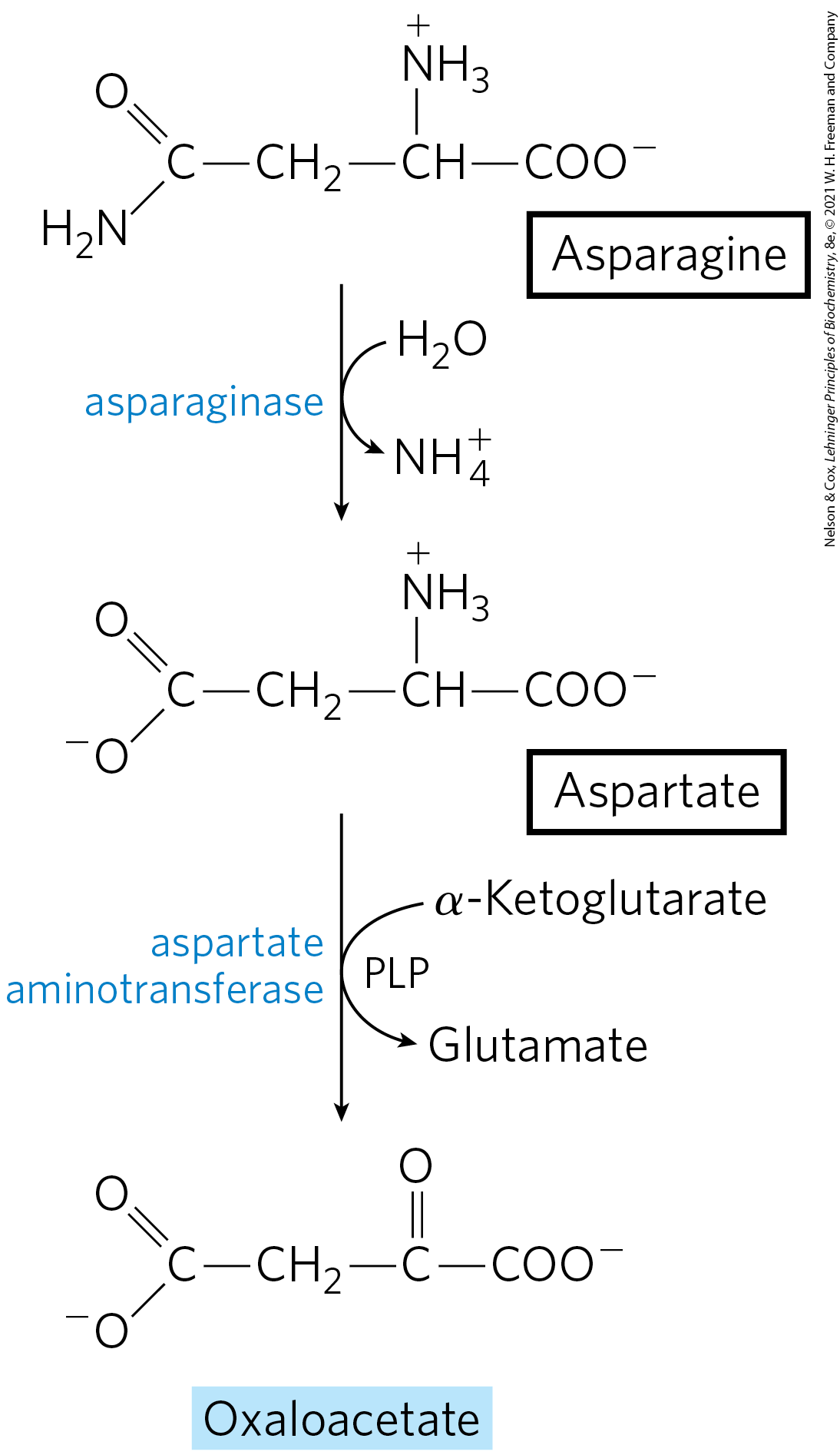
Asparagine and Aspartate Are Degraded to Oxaloacetate
The carbon skeletons of asparagine and aspartate ultimately enter the citric acid cycle as malate in mammals or oxaloacetate in bacteria. The enzyme asparaginase catalyzes the hydrolysis of asparagine to aspartate, which undergoes transamination with α-ketoglutarate to yield glutamate and oxaloacetate (Fig. 18-29). The oxaloacetate is converted to malate in the cytosol and then transported into the mitochondrial matrix through the malate–α-ketoglutarate transporter. In bacteria, the oxaloacetate produced in the transamination reaction can be used directly in the citric acid cycle.
FIGURE 18-29 Catabolic pathway for asparagine and aspartate. Both amino acids are converted to oxaloacetate.
We have now seen how the 20 common amino acids, after losing their nitrogen atoms, are degraded by dehydrogenation, decarboxylation, and other reactions to yield portions of their carbon backbones in the form of six central metabolites that can enter the citric acid cycle. Those portions degraded to acetyl-CoA are completely oxidized to carbon dioxide and water, with generation of ATP by oxidative phosphorylation. Those portions degraded to other citric acid cycle intermediates can either be oxidized or contribute to gluconeogenesis, depending on the metabolic state.
As was the case for carbohydrates and lipids, the degradation of amino acids results ultimately in the generation of reducing equivalents (NADH and ) through the action of the citric acid cycle. Our survey of catabolic processes concludes in the next chapter with a discussion of respiration, in which these reducing equivalents fuel the ultimate oxidative and energy-generating process in aerobic organisms.
SUMMARY 18.3 Pathways of Amino Acid Degradation
After the removal of amino groups, the carbon skeletons of amino acids undergo oxidation to compounds that can enter the citric acid cycle. The citric acid cycle products can be oxidized as fuel. Alternatively, depending on their degradative end product, some amino acids can be converted to ketone bodies, some to glucose, and some to both. Thus, amino acid degradation is integrated into intermediary metabolism and can be critical to survival under conditions in which amino acids are a significant source of metabolic energy.
The reactions of these pathways require several cofactors, including tetrahydrofolate and S-adenosylmethionine in one-carbon transfer reactions and tetrahydrobiopterin in the oxidation of phenylalanine by phenylalanine hydroxylase.
Alanine, cysteine, glycine, serine, threonine, and tryptophan are converted in whole or in part to pyruvate.
Leucine, lysine, phenylalanine, tyrosine, and tryptophan yield acetyl-CoA via acetoacetyl-CoA. Isoleucine, leucine, threonine, and tryptophan also form acetyl-CoA directly. Leucine and lysine are the only amino acids that cannot contribute to gluconeogenesis. Four carbon atoms of phenylalanine and tyrosine give rise to fumarate.
Many human genetic diseases are traced to deficiencies in enzymes catalyzing steps in amino acid degradation. Two well-studied examples, phenylketonuria and alkaptonuria, feature defects in phenylalanine degradation. Most amino acid degradation deficiencies are treated with dietary intervention.
Arginine, glutamate, glutamine, histidine, and proline are degraded to α-ketoglutarate.
Isoleucine, methionine, threonine, and valine produce succinyl-CoA.
The branched-chain amino acids (isoleucine, leucine, and valine), unlike the other amino acids, are degraded only in extrahepatic tissues.
 The seven amino acids that are degraded entirely or in part to acetoacetyl-CoA and/or acetyl-CoA — phenylalanine, tyrosine, isoleucine, leucine, tryptophan, threonine, and lysine — can yield ketone bodies in the liver, where acetoacetyl-CoA is converted to acetoacetate and then to acetone and β-hydroxybutyrate (see
The seven amino acids that are degraded entirely or in part to acetoacetyl-CoA and/or acetyl-CoA — phenylalanine, tyrosine, isoleucine, leucine, tryptophan, threonine, and lysine — can yield ketone bodies in the liver, where acetoacetyl-CoA is converted to acetoacetate and then to acetone and β-hydroxybutyrate (see 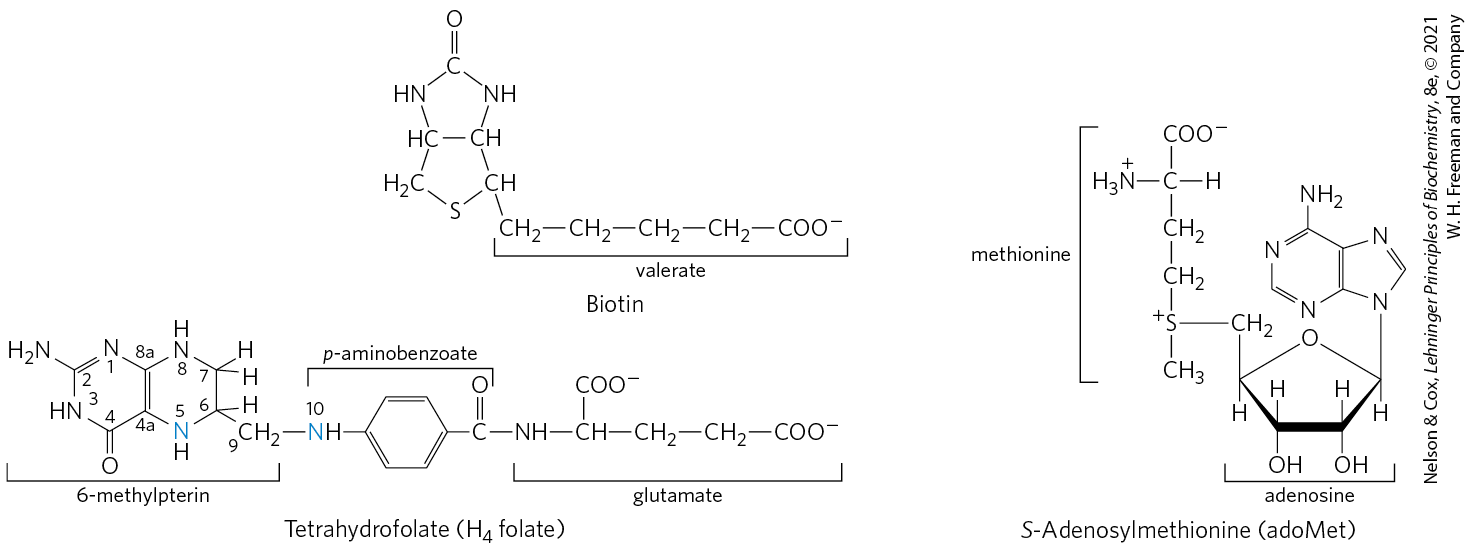
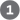 ). This reaction is unusual in that the nucleophilic sulfur atom of methionine attacks the carbon of the ribose moiety of ATP rather than one of the phosphorus atoms. Triphosphate is released and is cleaved to and on the enzyme, and the is cleaved by inorganic pyrophosphatase; thus three bonds, including two bonds of high-energy phosphate groups, are broken in this reaction. The only other known reaction in which triphosphate is displaced from ATP occurs in the synthesis of coenzyme (see
). This reaction is unusual in that the nucleophilic sulfur atom of methionine attacks the carbon of the ribose moiety of ATP rather than one of the phosphorus atoms. Triphosphate is released and is cleaved to and on the enzyme, and the is cleaved by inorganic pyrophosphatase; thus three bonds, including two bonds of high-energy phosphate groups, are broken in this reaction. The only other known reaction in which triphosphate is displaced from ATP occurs in the synthesis of coenzyme (see 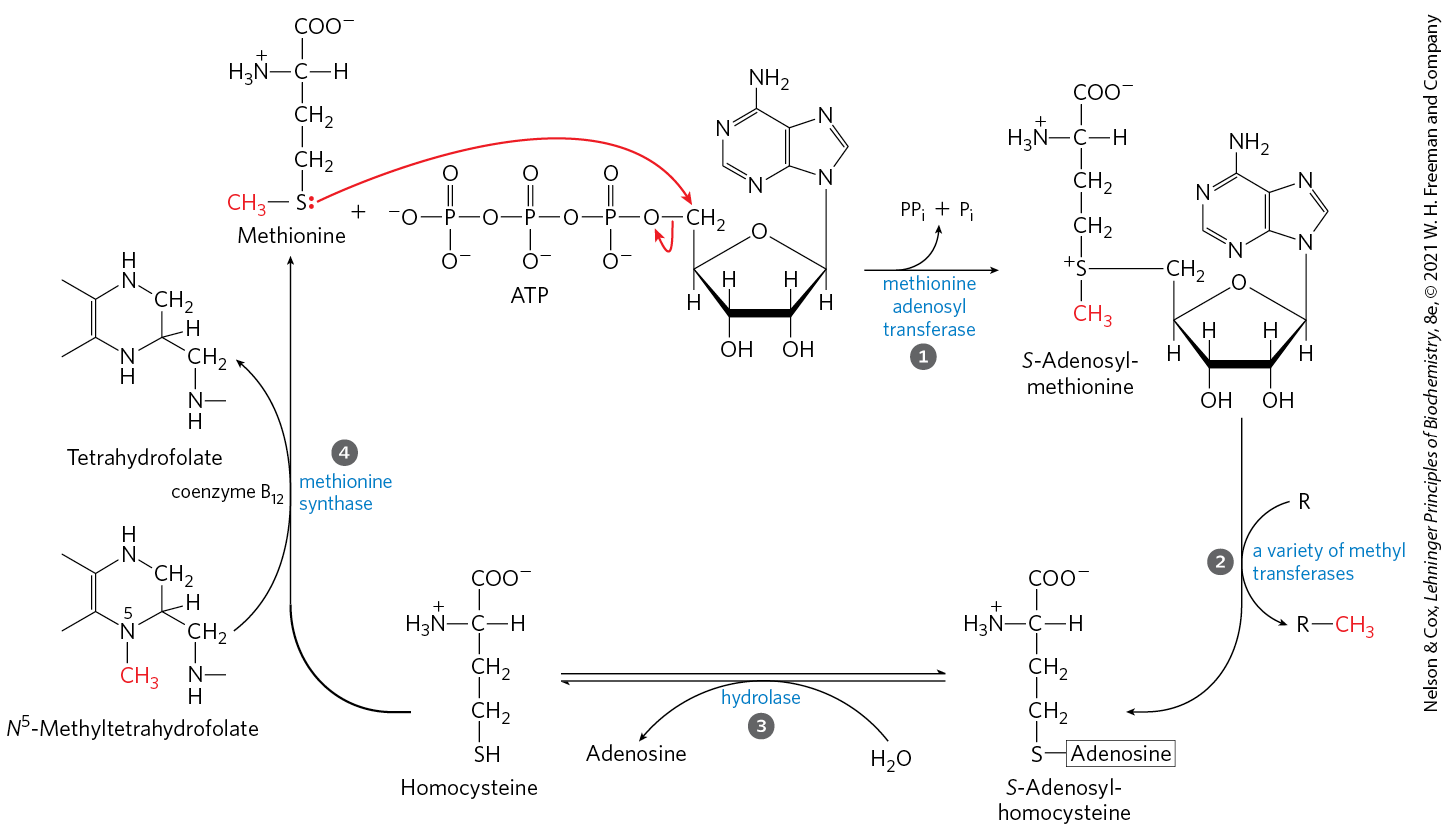
 ), the methyl group is transferred to cobalamin to form methylcobalamin, which is the methyl donor in the formation of methionine. S-Adenosylmethionine, which has a positively charged sulfur (and is thus a sulfonium ion), is a powerful methylating agent in several biosynthetic reactions. The methyl group acceptor (step
), the methyl group is transferred to cobalamin to form methylcobalamin, which is the methyl donor in the formation of methionine. S-Adenosylmethionine, which has a positively charged sulfur (and is thus a sulfonium ion), is a powerful methylating agent in several biosynthetic reactions. The methyl group acceptor (step  ) is designated R.
) is designated R. ). Methionine is regenerated by transfer of a methyl group to homocysteine in a reaction catalyzed by methionine synthase (step
). Methionine is regenerated by transfer of a methyl group to homocysteine in a reaction catalyzed by methionine synthase (step  The vitamins and folate are closely linked in these pathways. The anemia observed in the rare deficiency disease pernicious anemia (
The vitamins and folate are closely linked in these pathways. The anemia observed in the rare deficiency disease pernicious anemia (



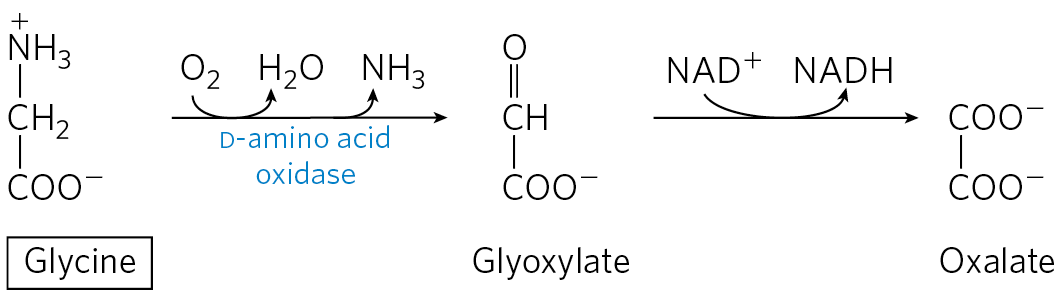

 After the removal of amino groups, the carbon skeletons of amino acids undergo oxidation to compounds that can enter the citric acid cycle. The citric acid cycle products can be oxidized as fuel. Alternatively, depending on their degradative end product, some amino acids can be converted to ketone bodies, some to glucose, and some to both. Thus, amino acid degradation is integrated into intermediary metabolism and can be critical to survival under conditions in which amino acids are a significant source of metabolic energy.
After the removal of amino groups, the carbon skeletons of amino acids undergo oxidation to compounds that can enter the citric acid cycle. The citric acid cycle products can be oxidized as fuel. Alternatively, depending on their degradative end product, some amino acids can be converted to ketone bodies, some to glucose, and some to both. Thus, amino acid degradation is integrated into intermediary metabolism and can be critical to survival under conditions in which amino acids are a significant source of metabolic energy.