As discussed in Chapter 8, nucleotides have a variety of important functions in all cells. They are the precursors of DNA and RNA. They are essential carriers of chemical energy — a role primarily of ATP and to some extent GTP. They are components of the cofactors NAD, FAD, S-adenosylmethionine, and coenzyme A, as well as of activated biosynthetic intermediates such as UDP-glucose and CDP-diacylglycerol. Some, such as cAMP and cGMP, are also cellular second messengers.
Two types of pathways lead to nucleotides: the de novo pathways and the salvage pathways. De novo synthesis of nucleotides begins with their metabolic precursors: amino acids, ribose 5-phosphate, , and . Salvage pathways recycle the free bases and nucleosides released from nucleic acid breakdown. Both types of pathways are important in cellular metabolism, and both are discussed in this section.
The de novo pathways for purine and pyrimidine biosynthesis seem to be nearly identical in all living organisms. Notably, the free bases guanine, adenine, thymine, cytidine, and uracil are not intermediates in these pathways; that is, the bases are not synthesized and then attached to ribose, as might be expected. The purine ring structure is built up one or a few atoms at a time, attached to ribose throughout the process. The pyrimidine ring is synthesized as orotate, attached to ribose phosphate, and then converted to the common pyrimidine nucleotides required in nucleic acid synthesis. Although the free bases are not intermediates in the de novo pathways, they are intermediates in some of the salvage pathways.
Several important precursors are shared by the de novo pathways for synthesis of pyrimidines and purines. Phosphoribosyl pyrophosphate (PRPP) is important in both, and in these pathways the structure of ribose is retained in the product nucleotide, in contrast to its fate in the tryptophan and histidine biosynthetic pathways discussed earlier. An amino acid is an important precursor in each type of pathway: glycine for purines and aspartate for pyrimidines. Glutamine again is the most important source of amino groups — in five different steps in the de novo pathways. Aspartate is also used as the source of an amino group in the purine pathways, in two steps.
Two other features deserve mention. First, there is evidence, especially in the de novo purine pathway, that the enzymes are present as large, multienzyme complexes or metabolons in the cell, a recurring theme in our discussion of metabolism. Second, the cellular pools of nucleotides (other than ATP) are quite small, perhaps 1% or less of the amounts required to synthesize the cell’s DNA. Therefore, cells must continue to synthesize nucleotides during nucleic acid synthesis, and in some cases, nucleotide synthesis may limit the rates of DNA replication and transcription. Because of the importance of these processes in dividing cells, agents that inhibit nucleotide synthesis have become particularly important in medicine.
We examine here the biosynthetic pathways of purine and pyrimidine nucleotides and their regulation, the formation of the deoxynucleotides, and the degradation of purines and pyrimidines to uric acid and urea. We end with a discussion of chemotherapeutic agents that affect nucleotide synthesis.
De Novo Purine Nucleotide Synthesis Begins with PRPP
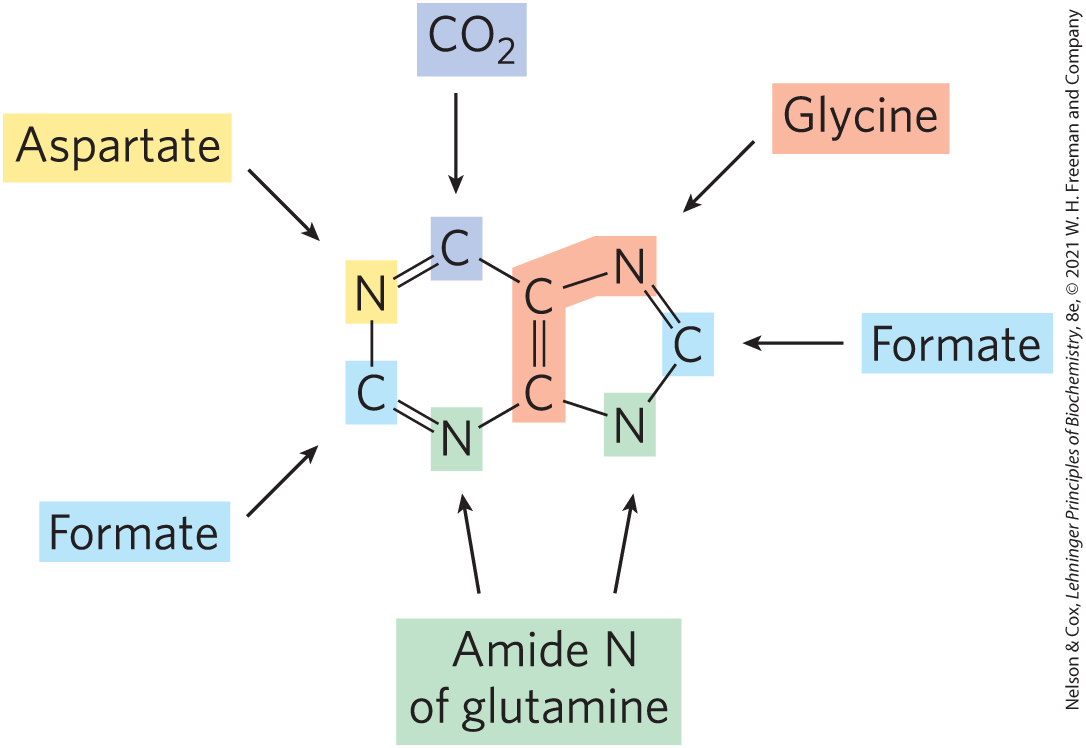
The two parent purine nucleotides of nucleic acids are adenosine -monophosphate (AMP; adenylate) and guanosine -monophosphate (GMP; guanylate), containing the purine bases adenine and guanine. Figure 22-34 shows the origin of the carbon and nitrogen atoms of the purine ring system, as determined by John M. Buchanan using isotopic tracer experiments in birds (who conveniently excrete excess nitrogen as insoluble uric acid, a purine analog). The detailed pathway of purine biosynthesis was worked out primarily by Buchanan and G. Robert Greenberg in the 1950s.
FIGURE 22-34 Origin of the ring atoms of purines. This information was obtained from isotopic experiments with - or -labeled precursors. Formate is supplied in the form of -formyltetrahydrofolate.
In the first committed step of the pathway, an amino group donated by glutamine is attached at C-1 of PRPP (Fig. 22-35). The resulting 5-phosphoribosylamine is highly unstable, with a half-life of 30 seconds at pH 7.5. This intermediate is rapidly funneled into the next biosynthetic step, and the purine ring is subsequently built up on this structure. The pathway described here is identical in all organisms, with the exception of one step that differs in higher eukaryotes, as noted below.
FIGURE 22-35 De novo synthesis of purine nucleotides: construction of the purine ring of inosinate (IMP). Each addition to the purine ring is shaded to match Figure 22-34. After step , R symbolizes the 5-phospho-d-ribosyl group on which the purine ring is built. Formation of 5-phosphoribosylamine (step ) is the first committed step in purine synthesis. Note that the product of step , AICAR, is the remnant of ATP released during histidine biosynthesis (see Fig. 22-22, step ). Abbreviations are given for most intermediates to simplify the naming of the enzymes. Step is the alternative path from AIR to CAIR occurring in higher eukaryotes.
The second step is the addition of three atoms from glycine (Fig. 22-35, step ). An ATP is consumed to activate the glycine carboxyl group (in the form of an acyl phosphate) for this condensation reaction. The added glycine amino group is then formylated by -formyltetrahydrofolate (step ), and a nitrogen is contributed by glutamine (step ), before dehydration and ring closure yield the five-membered imidazole ring of the purine nucleus, as 5-aminoimidazole ribonucleotide (AIR; step ).
At this point, three of the six atoms needed for the second ring in the purine structure are in place. To complete the process, a carboxyl group is first added (step ). This carboxylation is unusual in that it does not require biotin, but instead uses the bicarbonate generally present in aqueous solutions. A rearrangement transfers the carboxylate from the exocyclic amino group to position 4 of the imidazole ring (step ). Steps and are found only in bacteria and fungi. In higher eukaryotes, including humans, the 5-aminoimidazole ribonucleotide product of step is carboxylated directly to carboxyaminoimidazole ribonucleotide in one step instead of two (step ). The enzyme catalyzing this reaction is AIR carboxylase.
Aspartate now donates its amino group in two steps ( and ): formation of an amide bond, followed by elimination of the carbon skeleton of aspartate (as fumarate). (Recall that aspartate plays an analogous role in two steps of the urea cycle; see Fig. 18-10.) The final carbon is contributed by -formyltetrahydrofolate (step ), and a second ring closure takes place to yield the second fused ring of the purine nucleus (step ). The first intermediate with a complete purine ring is inosinate (IMP).
As in the tryptophan and histidine biosynthetic pathways, the enzymes of IMP synthesis seem to be organized as large metabolons in the cell. Once again, evidence comes from the existence of single polypeptides with several functions, some catalyzing nonsequential steps in the pathway. In eukaryotic cells ranging from yeast to fruit flies to chickens, steps , , and in Figure 22-35 are catalyzed by a multifunctional protein. An additional multifunctional protein catalyzes steps and . In humans, a multifunctional enzyme combines the activities of AIR carboxylase and SAICAR synthetase (steps and ). In bacteria, these activities are found on separate proteins, but the proteins may form a metabolon. The channeling of reaction intermediates from one enzyme to the next permitted by these complexes is probably especially important for unstable intermediates such as 5-phosphoribosylamine.
Conversion of inosinate to adenylate requires the insertion of an amino group derived from aspartate (Fig. 22-36); this takes place in two reactions similar to those used to introduce N-1 of the purine ring (Fig. 22-35, steps and ). A crucial difference is that GTP rather than ATP is the source of the high-energy phosphate in synthesizing adenylosuccinate. Guanylate is formed by the -requiring oxidation of inosinate at C-2, followed by addition of an amino group derived from glutamine. ATP is cleaved to AMP and in the final step (Fig. 22-36).
FIGURE 22-36 Biosynthesis of AMP and GMP from IMP.
Purine Nucleotide Biosynthesis Is Regulated by Feedback Inhibition
Four major feedback mechanisms cooperate in regulating the overall rate of de novo purine nucleotide synthesis and the relative rates of formation of the two end products, adenylate and guanylate (Fig. 22-37). The first mechanism is exerted on the first reaction that is unique to purine synthesis: transfer of an amino group to PRPP to form 5-phosphoribosylamine. This reaction is catalyzed by the allosteric enzyme glutamine-PRPP amidotransferase, which is inhibited by the end products IMP, AMP, and GMP. AMP and GMP act synergistically in this concerted inhibition. Thus, whenever either AMP or GMP accumulates to excess, the first step in its biosynthesis from PRPP is partially inhibited.
FIGURE 22-37 Regulatory mechanisms in the biosynthesis of adenine and guanine nucleotides in E. coli. Regulation of these pathways differs in other organisms.
In the second control mechanism, exerted at a later stage, an excess of GMP in the cell inhibits formation of xanthylate from inosinate by IMP dehydrogenase, without affecting the formation of AMP. Conversely, an accumulation of adenylate inhibits formation of adenylosuccinate by adenylosuccinate synthetase, without affecting the biosynthesis of GMP. When both products are present in sufficient quantities, IMP builds up, and it inhibits an earlier step in the pathway; this is another example of the regulatory strategy called sequential feedback inhibition.
In the third mechanism, GTP is required in the conversion of IMP to AMP, whereas ATP is required for conversion of IMP to GMP (Fig. 22-36), a reciprocal arrangement that tends to balance the synthesis of the two ribonucleotides.
The fourth and final control mechanism is the inhibition of PRPP synthesis by the allosteric regulation of ribose phosphate pyrophosphokinase. This enzyme is inhibited by ADP and GDP, in addition to metabolites from other pathways for which PRPP is a starting point.
Pyrimidine Nucleotides Are Made from Aspartate, PRPP, and Carbamoyl Phosphate
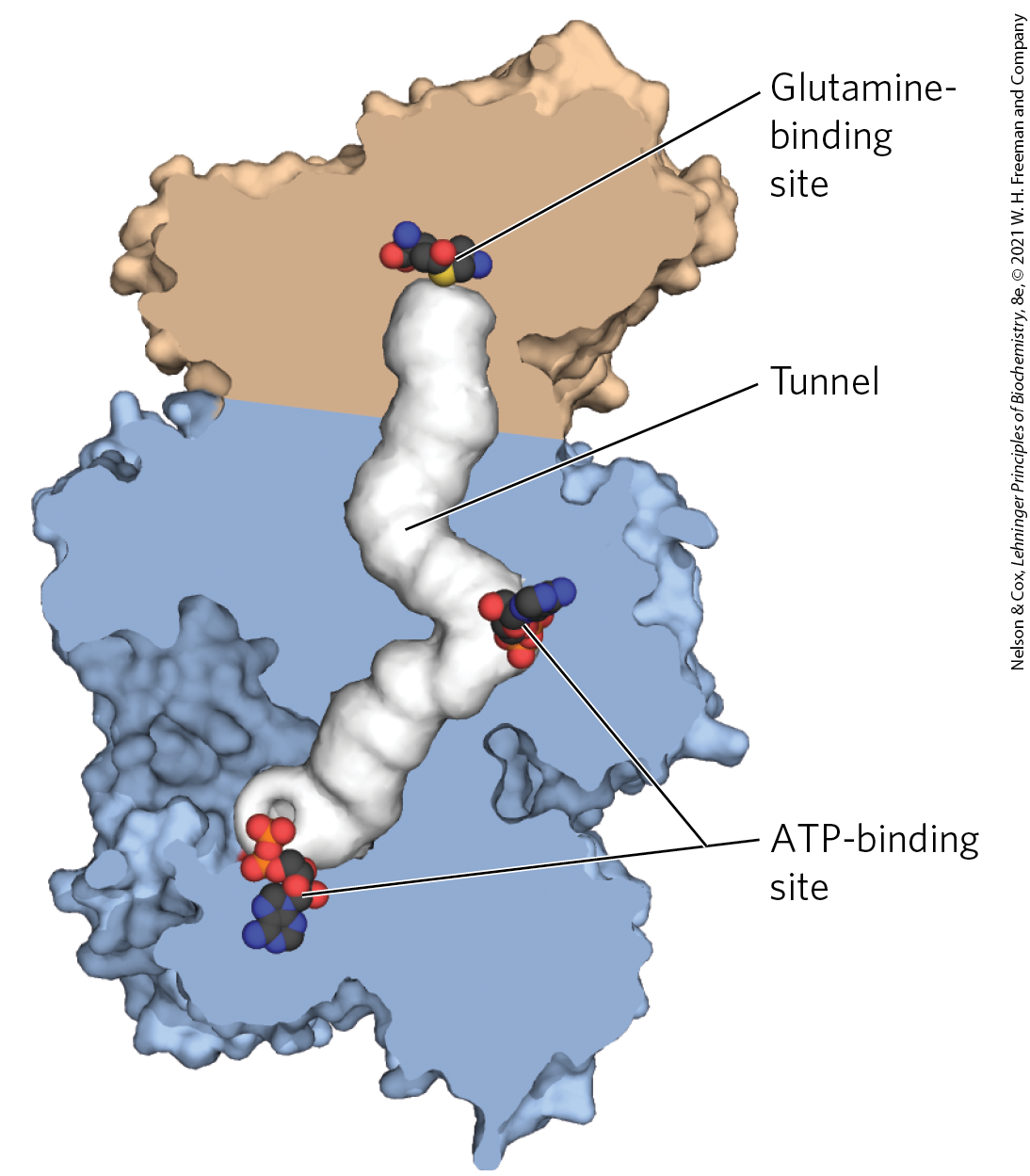
The common pyrimidine ribonucleotides are cytidine -monophosphate (CMP; cytidylate) and uridine -monophosphate (UMP; uridylate), which contain the pyrimidines cytosine and uracil. De novo pyrimidine nucleotide biosynthesis (Fig. 22-38) proceeds in a somewhat different manner from purine nucleotide synthesis; the six-membered pyrimidine ring is made first and then attached to ribose 5-phosphate. Required in this process is carbamoyl phosphate, also an intermediate in the urea cycle. However, in animals the carbamoyl phosphate required in urea synthesis is made in mitochondria by carbamoyl phosphate synthetase I, whereas the carbamoyl phosphate required in pyrimidine biosynthesis is made in the cytosol by a different form of the enzyme, carbamoyl phosphate synthetase II. In bacteria, a single enzyme supplies carbamoyl phosphate for the synthesis of arginine and pyrimidines. The bacterial enzyme has three separate active sites, spaced along a channel nearly 100 Å long (Fig. 22-39). Bacterial carbamoyl phosphate synthetase provides a vivid illustration of the channeling of unstable reaction intermediates between active sites so that products are formed efficiently.
FIGURE 22-38 De novo synthesis of pyrimidine nucleotides: biosynthesis of UTP and CTP via orotidylate. The pyrimidine is constructed from carbamoyl phosphate and aspartate. The ribose 5-phosphate is then added to the completed pyrimidine ring by orotate phosphoribosyltransferase. The first step in this pathway (not shown here; see Fig. 18-11a) is the synthesis of carbamoyl phosphate from , , and ATP. In eukaryotes, the first step is catalyzed by carbamoyl phosphate synthetase II.
FIGURE 22-39 Channeling of intermediates in bacterial carbamoyl phosphate synthetase. The reaction catalyzed by this enzyme (and its mitochondrial counterpart) is illustrated in Figure 18-11a. In this cutaway, the small and large subunits are shown in tan and blue, respectively; the tunnel between active sites (almost 100 Å long) is shown as white. In this reaction, a glutamine molecule binds to the small subunit, donating its amido nitrogen as in a glutamine amidotransferase–type reaction. The enters the tunnel, which takes it to a second active site, where it combines with bicarbonate in a reaction requiring ATP. The carbamate then reenters the tunnel to reach the third active site, where it is phosphorylated by ATP to carbamoyl phosphate. To solve this structure, the enzyme was crystallized with ornithine bound to the glutamine-binding site and ADP bound to the ATP-binding sites. [Data from PDB ID 1M6V, J. B. Thoden et al., J. Biol. Chem. 277:39,722, 2002.]
Carbamoyl phosphate reacts with aspartate to yield N-carbamoylaspartate in the first committed step of pyrimidine biosynthesis (Fig. 22-38). This reaction is catalyzed by aspartate transcarbamoylase. In bacteria, this step is highly regulated, and bacterial aspartate transcarbamoylase is one of the most thoroughly studied allosteric enzymes (see below). By removal of water from N-carbamoylaspartate, a reaction catalyzed by dihydroorotase, the pyrimidine ring is closed to form l-dihydroorotate. This compound is oxidized to the pyrimidine derivative orotate, a reaction in which is the ultimate electron acceptor. In eukaryotes, the first three enzymes in this pathway — carbamoyl phosphate synthetase II, aspartate transcarbamoylase, and dihydroorotase — are part of a single trifunctional protein. The protein, known by the acronym CAD, contains three identical polypeptide chains (each of ), each with active sites for all three reactions. This suggests that metabolons may be the rule in this pathway.
Once orotate is formed, the ribose 5-phosphate side chain, provided once again by PRPP, is attached to yield orotidylate (Fig. 22-38). Orotidylate is then decarboxylated to uridylate, which is phosphorylated to UTP. CTP is formed from UTP by the action of cytidylate synthetase, by way of an acyl phosphate intermediate (consuming one ATP). The nitrogen donor is normally glutamine, although the cytidylate synthetases in many species can use directly.
Pyrimidine Nucleotide Biosynthesis Is Regulated by Feedback Inhibition
Regulation of the rate of pyrimidine nucleotide synthesis in bacteria occurs in large part through aspartate transcarbamoylase (ATCase), which catalyzes the first reaction in the sequence and is inhibited by CTP, the end product of the sequence (Fig. 22-38). The bacterial ATCase molecule consists of six catalytic subunits and six regulatory subunits (see Fig. 6-36). The catalytic subunits bind the substrate molecules, and the allosteric subunits bind the allosteric inhibitor, CTP. The entire ATCase molecule, as well as its subunits, exists in two conformations, active and inactive. When CTP is not bound to the regulatory subunits, the enzyme is maximally active. As CTP accumulates and binds to the regulatory subunits, they undergo a change in conformation. This change is transmitted to the catalytic subunits, which then also shift to an inactive conformation. ATP prevents the changes induced by CTP. Figure 22-40 shows the effects of the allosteric regulators on the activity of ATCase.
FIGURE 22-40 Allosteric regulation of aspartate transcarbamoylase by CTP and ATP. Addition of CTP, the allosteric inhibitor of ATCase, increases the for aspartate (lower curve), thereby reducing the rate of conversion of aspartate to N-carbamoylaspartate. ATP at fully reverses this inhibition by CTP (middle curve).
Nucleoside Monophosphates Are Converted to Nucleoside Triphosphates
Nucleotides to be used in biosynthesis are generally converted to nucleoside triphosphates. The conversion pathways are common to all cells. Phosphorylation of AMP to ADP is promoted by adenylate kinase, in the reaction
The ADP so formed is phosphorylated to ATP by the glycolytic enzymes or through oxidative phosphorylation.
ATP also brings about the formation of other nucleoside diphosphates by the action of a class of enzymes called nucleoside monophosphate kinases. These enzymes, which are generally specific for a particular base but nonspecific for the sugar (ribose or deoxyribose), catalyze the reaction
The efficient cellular systems for rephosphorylating ADP to ATP (ATP synthase; Chapter 19) tend to remove ADP and pull this reaction in the direction of products.
Nucleoside diphosphates are converted to triphosphates by the action of a ubiquitous enzyme, nucleoside diphosphate kinase, which catalyzes the reaction
This enzyme is notable in that it is not specific for the base (purines or pyrimidines) or the sugar (ribose or deoxyribose). This nonspecificity applies to both phosphate acceptor (A) and donor (D), although the donor is almost invariably ATP because it is present in higher concentration than other nucleoside triphosphates under aerobic conditions.
Ribonucleotides Are the Precursors of Deoxyribonucleotides
Deoxyribonucleotides, the building blocks of DNA, are derived from the corresponding ribonucleotides by direct reduction at the -carbon atom of the d-ribose to form the -deoxy derivative. For example, adenosine diphosphate (ADP) is reduced to -deoxyadenosine diphosphate (dADP), and GDP is reduced to dGDP. This reaction is somewhat unusual in that the reduction occurs at a nonactivated carbon; no closely analogous chemical reactions are known. The reaction is catalyzed by ribonucleotide reductase, best characterized in E. coli, in which its substrates are ribonucleoside diphosphates.
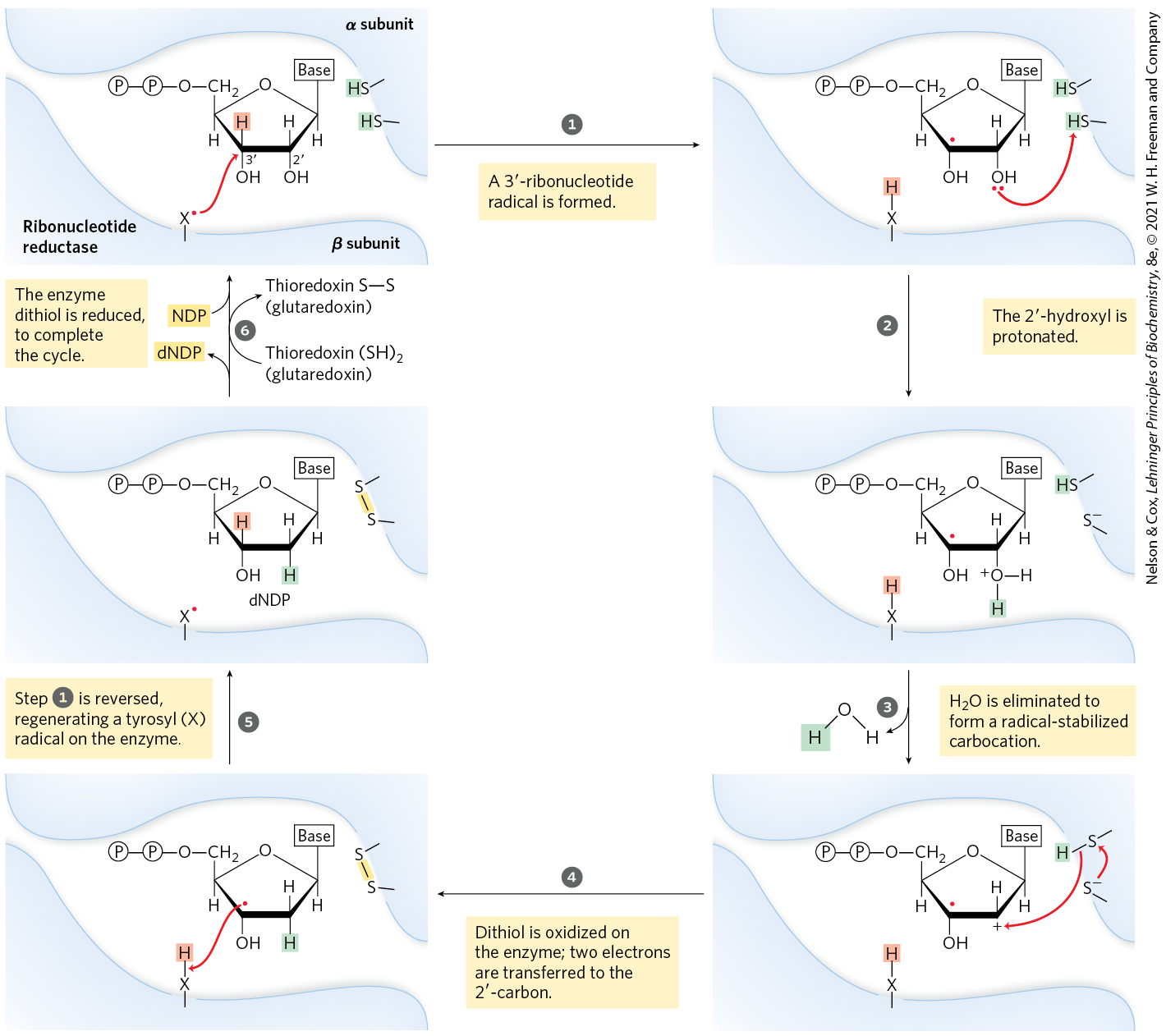
The reduction of the d-ribose portion of a ribonucleoside diphosphate to -deoxy-d-ribose requires a pair of hydrogen atoms, which are ultimately donated by NADPH via an intermediate hydrogen-carrying protein, thioredoxin. This ubiquitous protein serves a similar redox function in photosynthesis (see Fig. 20-37) and other processes. Thioredoxin has pairs of groups that carry hydrogen atoms from NADPH to the ribonucleoside diphosphate. Its oxidized (disulfide) form is reduced by NADPH in a reaction catalyzed by thioredoxin reductase (Fig. 22-41), and reduced thioredoxin is then used by ribonucleotide reductase to reduce the nucleoside diphosphates (NDPs) to deoxyribonucleoside diphosphates (dNDPs). A second source of reducing equivalents for ribonucleotide reductase is glutathione (GSH). Glutathione serves as the reductant for a protein closely related to thioredoxin, glutaredoxin, which then transfers the reducing power to ribonucleotide reductase.
FIGURE 22-41 Reduction of ribonucleotides to deoxyribonucleotides by ribonucleotide reductase. Electrons are transmitted (red arrows) to the enzyme from NADPH via (a) glutaredoxin or (b) thioredoxin. The sulfhydryl groups in glutaredoxin reductase are contributed by two molecules of bound glutathione (GSH; GSSG indicates oxidized glutathione). Note that thioredoxin reductase is a flavoenzyme, with FAD as a prosthetic group.
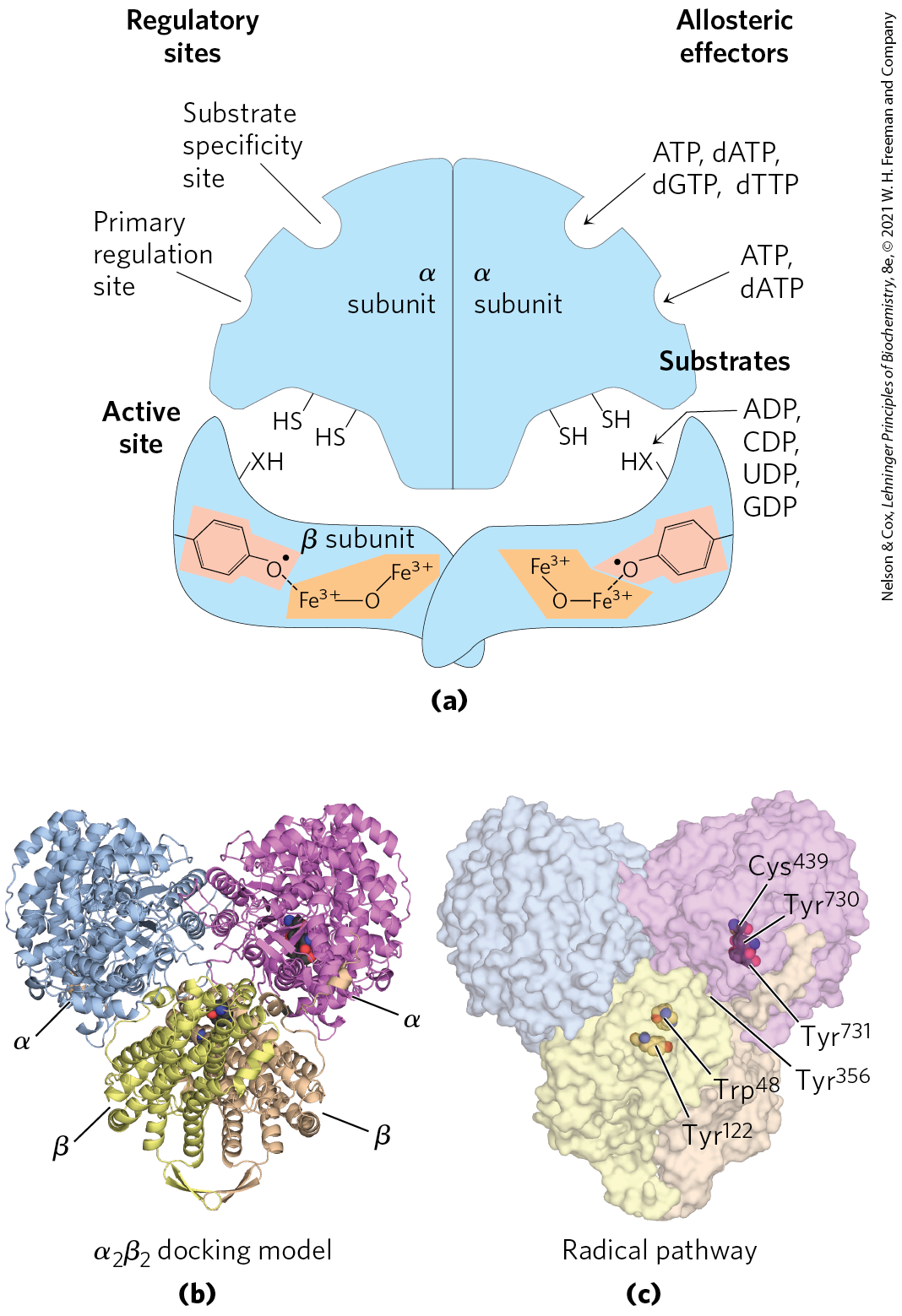
Ribonucleotide reductase is notable in that its reaction mechanism provides the best-characterized example of the involvement of free radicals in biochemical transformations, once thought to be rare in biological systems. The enzyme in E. coli and most eukaryotes is an dimer, with two catalytic subunits, , and two radical-generation subunits, (Fig. 22-42). Each catalytic subunit contains two kinds of regulatory sites, as described below. The two active sites of the enzyme are formed at the interface between the catalytic and radical-generation subunits. At each active site, an α subunit contributes two sulfhydryl groups required for activity, and the subunits contribute a stable tyrosyl radical. The subunits also have a binuclear iron cofactor that helps generate and stabilize the radical. The tyrosyl radical is too far from the active site to interact directly with the site, but several aromatic residues form a long-range radical-transfer pathway to the active site (Fig. 22-42c). A likely mechanism for the ribonucleotide reductase reaction is illustrated in Figure 22-43. In E. coli, the sources of the required reducing equivalents for this reaction are thioredoxin and glutaredoxin, as noted above.
FIGURE 22-42 Ribonucleotide reductase. (a) A schematic diagram of the subunit structures. Each catalytic subunit (α; also called R1) contains the two regulatory sites described in Figure 22-44 and two Cys residues central to the reaction mechanism. The radical-generation subunits (β; also called R2) each contain a critical residue and binuclear iron center. (b) The likely structure of . (c) The likely path of radical formation from the initial in a β subunit to the active-site , which is used in the mechanism shown in Figure 22-43. Several aromatic amino acid residues participate in long-range transfer of the radical from the point of its formation at to the active site, where the nucleotide substrate is bound. [(a) Information from L. Thelander and P. Reichard, Annu. Rev. Biochem. 48:133, 1979. (b, c) Data from PDB ID 3UUS, N. Ando et al., Proc. Natl. Acad. Sci. USA 108:21,046, 2011.]
MECHANISM FIGURE 22-43 Proposed mechanism for ribonucleotide reductase. In the enzyme of E. coli and most eukaryotes, the active thiol groups are on the α subunit. The active-site radical is on the β subunit and in E. coli is probably a thiyl radical of (see Fig. 22-42).
Three classes of ribonucleotide reductase have been reported. Their mechanisms (where known) generally conform to the scheme in Figure 22-43, but they differ in the identity of the group supplying the active-site radical and in the cofactors used to generate it. The E. coli enzyme (class I) requires oxygen to regenerate the tyrosyl radical if it is quenched, so this enzyme functions only in an aerobic environment. Class II enzymes, found in other microorganisms, have -deoxyadenosylcobalamin (see Box 17-2) rather than a binuclear iron center. Class III enzymes have evolved to function in an anaerobic environment. E. coli contains a separate class III ribonucleotide reductase when grown anaerobically; this enzyme contains an iron-sulfur cluster (structurally distinct from the binuclear iron center of the class I enzyme) and requires NADPH and S-adenosylmethionine for activity. It uses nucleoside triphosphates rather than nucleoside diphosphates as substrates. The evolution of different classes of ribonucleotide reductase for production of DNA precursors in different environments reflects the importance of this reaction in nucleotide metabolism.
Regulation of E. coli ribonucleotide reductase is unusual in that not only its activity but also its substrate specificity is regulated by the binding of effector molecules. Each α subunit has two types of regulatory sites (Fig. 22-42). One type affects overall enzyme activity and binds either ATP, which activates the enzyme, or dATP, which inactivates it. The second type alters substrate specificity in response to the effector molecule — ATP, dATP, dTTP, or dGTP — that is bound there (Fig. 22-44). When ATP or dATP is bound, reduction of UDP and CDP is favored. When dTTP or dGTP is bound, reduction of GDP or ADP, respectively, is stimulated. The scheme is designed to provide a balanced pool of precursors for DNA synthesis. ATP is also a general activator for biosynthesis and ribonucleotide reduction. The presence of dATP in small amounts increases the reduction of pyrimidine nucleotides. An oversupply of the pyrimidine dNTPs is signaled by high levels of dTTP. Abundant dTTP shifts the specificity to favor reduction of GDP. High levels of dGTP, in turn, shift the specificity to ADP reduction, and high levels of dATP shut the enzyme down. These effectors are thought to induce several distinct enzyme conformations with altered specificities.
FIGURE 22-44 Regulation of ribonucleotide reductase by deoxynucleoside triphosphates. The overall activity of the enzyme is affected by binding at the primary regulatory site (left). The substrate specificity of the enzyme is affected by the nature of the effector molecule bound at the second type of regulatory site, the substrate-specificity site (right). The diagram indicates inhibition or stimulation of enzyme activity with the four different substrates. The pathway from dUDP to dTMP is described below (see Figs 22-46, 22-47).
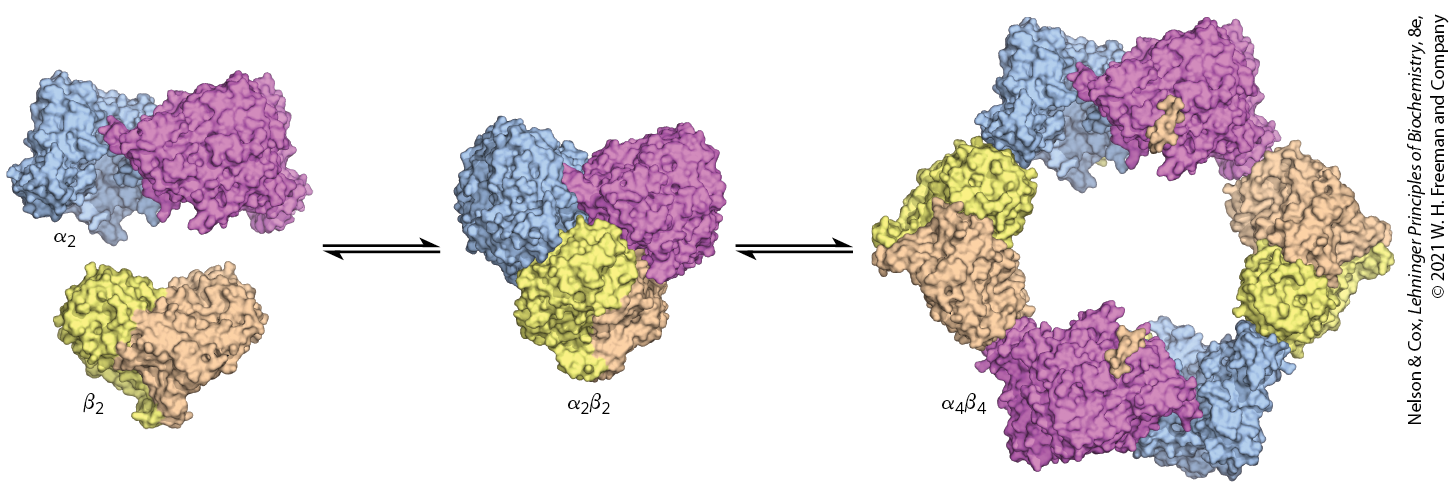
These regulatory effects are accompanied by, and presumably mediated by, large structural rearrangements in the enzyme. When the active form of the E. coli enzyme is inhibited by the addition of the allosteric inhibitor dATP, a ringlike structure forms, with alternating and subunits (Fig. 22-45). In this altered structure, the radical-forming path from β to α is disrupted and the residues in the path are exposed to solvent, effectively preventing radical transfer and thus inhibiting the reaction. The formation of ringlike structures is reversed when dATP levels are reduced. The yeast ribonucleotide reductase also undergoes oligomerization in the presence of dATP, forming a hexameric ring structure, .
FIGURE 22-45 Oligomerization of ribonucleotide reductase induced by the allosteric inhibitor dATP. At high concentrations of dATP , ring-shaped structures form. In this conformation, the residues in the radical-forming path are exposed to the solvent, blocking the radical reaction and inhibiting the enzyme. The oligomerization is reversed at lower dATP concentrations. [Data from PDB ID 3UUS, N. Ando et al., Proc. Natl. Acad. Sci. USA 108:21,046, 2011.]
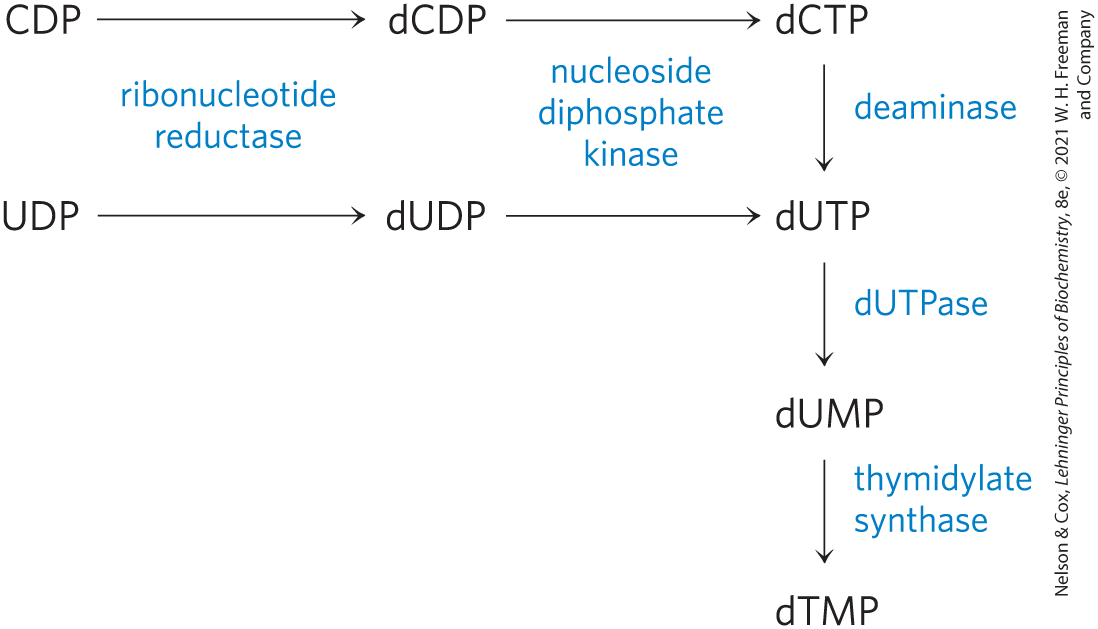
Thymidylate Is Derived from dCDP and dUMP
DNA contains thymine rather than uracil, and the de novo pathway to thymine involves only deoxyribonucleotides. The immediate precursor of thymidylate (dTMP) is dUMP. In bacteria, the pathway to dUMP begins with formation of dUTP, either by deamination of dCTP or by phosphorylation of dUDP (Fig. 22-46). The dUTP is converted to dUMP by a dUTPase. The latter reaction must be efficient to keep dUTP pools low and prevent incorporation of uridylate into DNA.
FIGURE 22-46 Biosynthesis of thymidylate (dTMP). The pathways are shown beginning with the reaction catalyzed by ribonucleotide reductase.
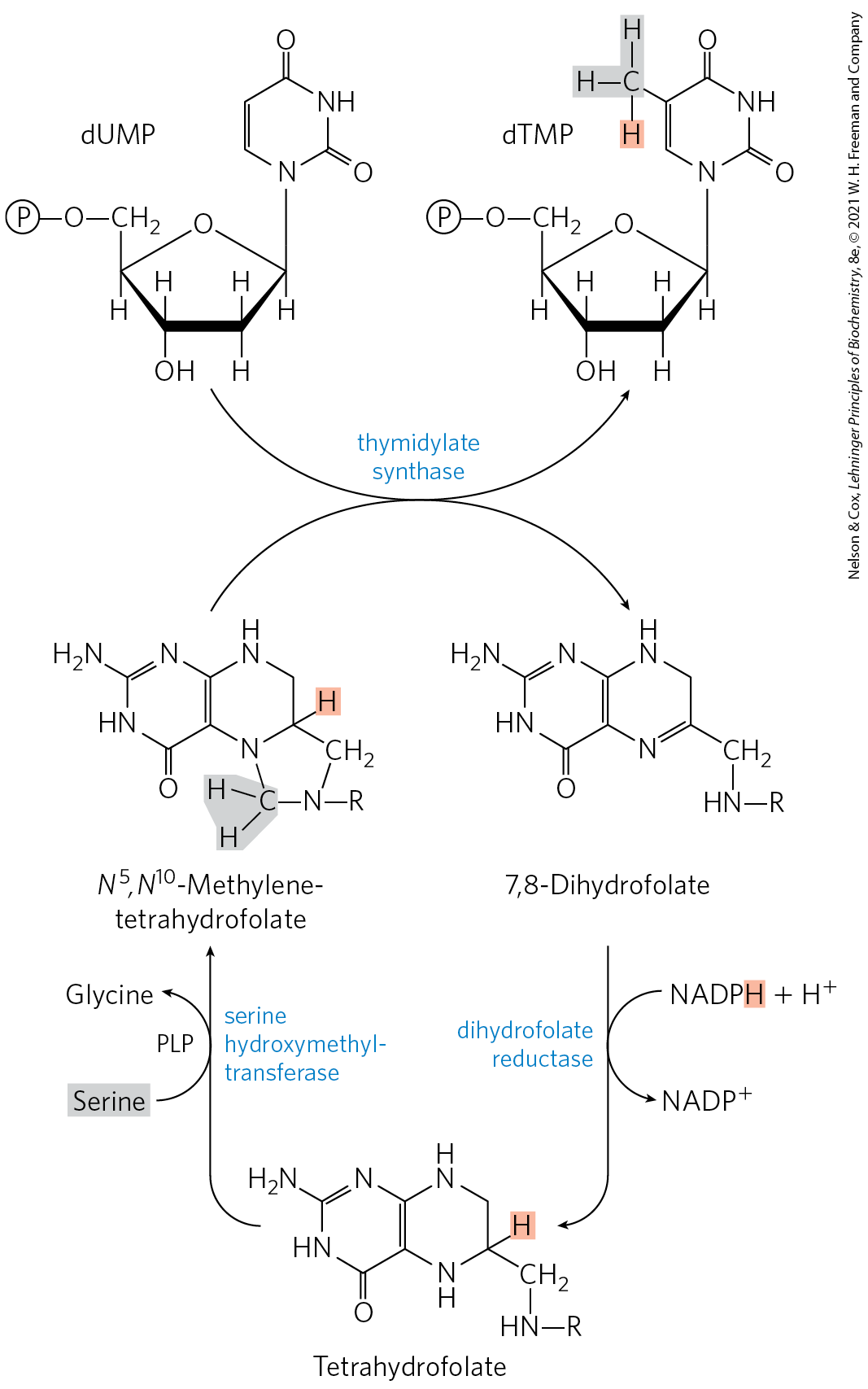
Conversion of dUMP to dTMP is catalyzed by thymidylate synthase. A one-carbon unit at the hydroxymethyl oxidation level (see Fig. 18-17) is transferred from -methylenetetrahydrofolate to dUMP, then reduced to a methyl group (Fig. 22-47). The reduction occurs at the expense of oxidation of tetrahydrofolate to dihydrofolate, which is unusual in tetrahydrofolate-requiring reactions. (The mechanism of this reaction is shown in Fig. 22-52.) The dihydrofolate is reduced to tetrahydrofolate by dihydrofolate reductase — a regeneration that is essential for the many processes that require tetrahydrofolate. In plants and at least one protist, thymidylate synthase and dihydrofolate reductase reside on a single, bifunctional protein.
FIGURE 22-47 Conversion of dUMP to dTMP by thymidylate synthase and dihydrofolate reductase. Serine hydroxymethyltransferase is required for regeneration of the -methylene form of tetrahydrofolate. In the synthesis of dTMP, all three hydrogens of the added methyl group are derived from -methylenetetrahydrofolate (light red and gray).
About 10% of the human population (and up to 50% of people in impoverished communities) suffers from folic acid deficiency. When the deficiency is severe, the symptoms can include heart disease, cancer, and some types of brain dysfunction. Folic acid deficiency during pregnancy can also produce neural tube defects in infants. At least some of these symptoms arise from a reduction in thymidylate synthesis, leading to an abnormal incorporation of uracil into DNA. Uracil is recognized by DNA repair pathways (described in Chapter 25) and is cleaved from the DNA. The presence of high levels of uracil in DNA leads to strand breaks that can greatly affect the function and regulation of nuclear DNA, ultimately causing the observed effects on the heart and brain, as well as increased mutagenesis that leads to cancer.
Degradation of Purines and Pyrimidines Produces Uric Acid and Urea, Respectively
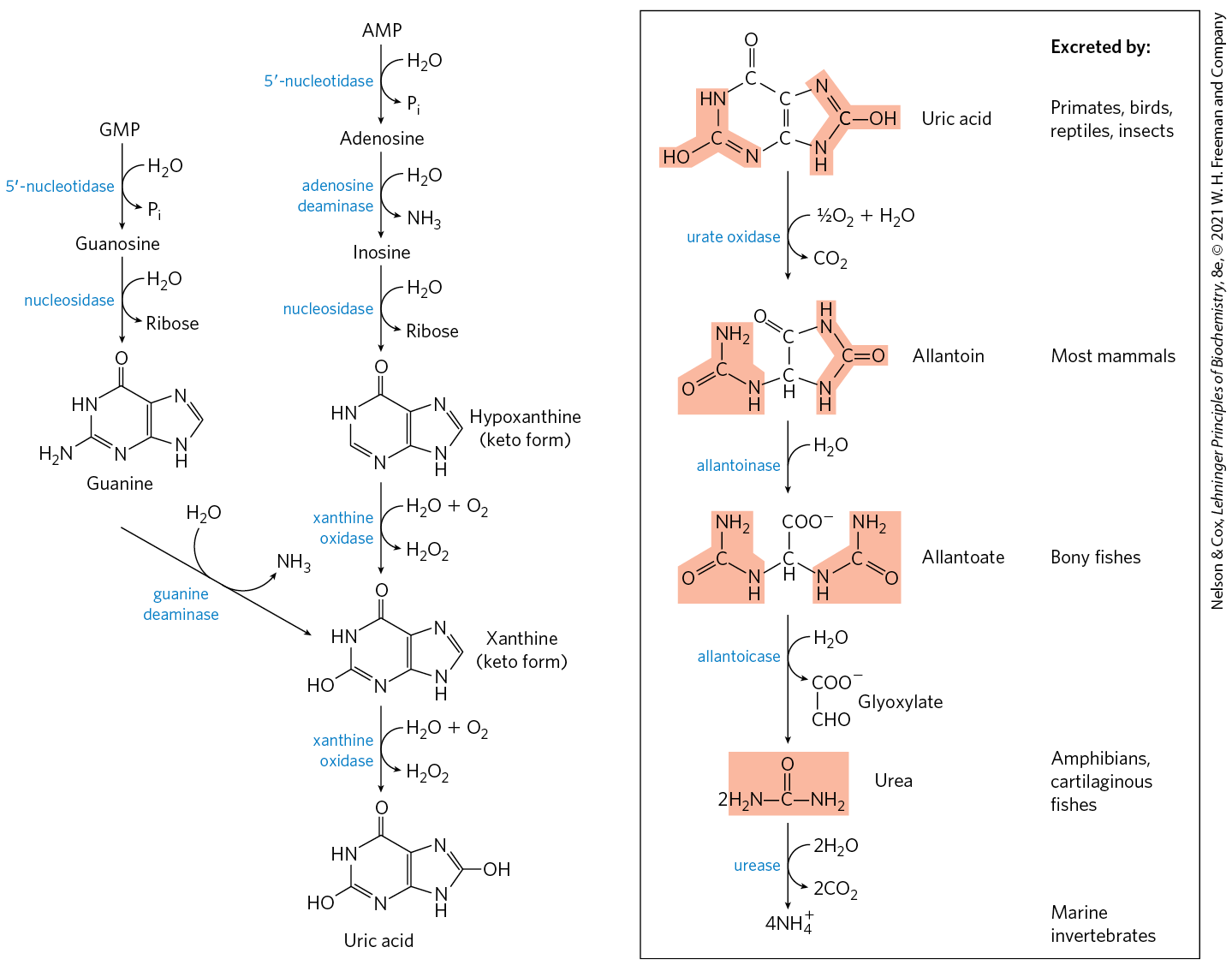
Purine nucleotides are degraded by a pathway in which they lose their phosphate through the action of -nucleotidase (Fig. 22-48). Adenylate yields adenosine, which is deaminated to inosine by adenosine deaminase, and inosine is hydrolyzed to hypoxanthine (its purine base) and d-ribose. Hypoxanthine is oxidized successively to xanthine and then uric acid by xanthine oxidase, a flavoenzyme with an atom of molybdenum and four iron-sulfur centers in its prosthetic group. Molecular oxygen is the electron acceptor in this complex reaction.
FIGURE 22-48 Catabolism of purine nucleotides. Note that primates excrete much more nitrogen as urea via the urea cycle (Chapter 18) than as uric acid from purine degradation. Similarly, fish excrete much more nitrogen as than as urea produced by the pathway shown here.
GMP catabolism also yields uric acid as an end product. GMP is first hydrolyzed to guanosine, which is then cleaved to free guanine. Guanine undergoes hydrolytic removal of its amino group to yield xanthine, which is converted to uric acid by xanthine oxidase.
Uric acid is the excreted end product of purine catabolism in primates, birds, and some other animals. A healthy adult human excretes uric acid at a rate of about 0.6 g/24 h; the excreted product arises in part from ingested purines and in part from turnover of the purine nucleotides of nucleic acids. In most mammals and many other vertebrates, uric acid is degraded to allantoin by the action of urate oxidase. In other organisms the pathway is further extended, as shown in Figure 22-48.
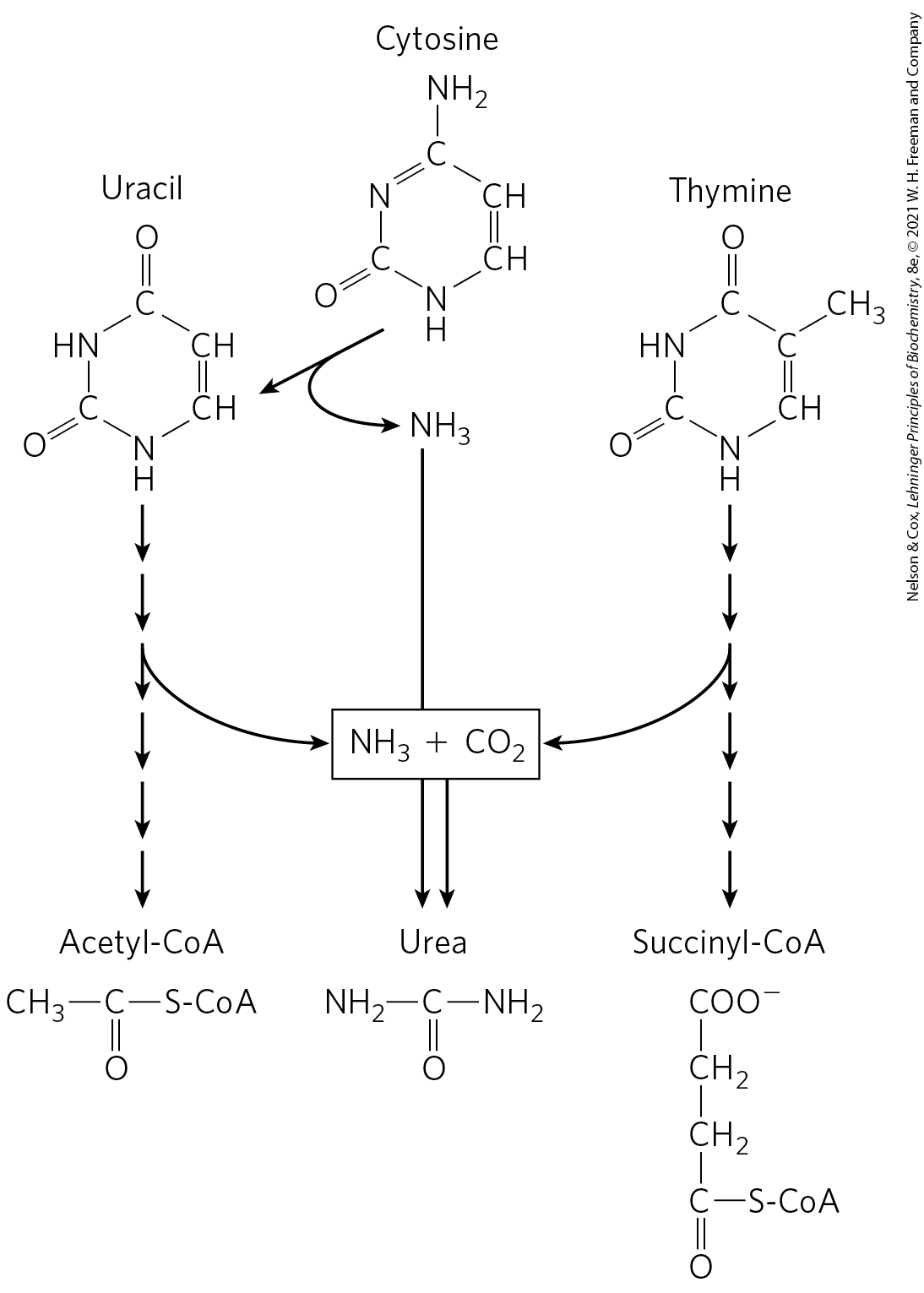
The pathways for degradation of pyrimidines generally lead to production and thus to urea synthesis. The carbons of thymine are degraded to succinyl-CoA; those of cytosine and uracil are degraded to acetyl-CoA (Fig. 22-49).
FIGURE 22-49 Catabolism of pyrimidines. These simplified pathways show end products but no intermediates.
Genetic aberrations in human purine metabolism have been found, some with serious consequences. For example, adenosine deaminase (ADA) deficiency leads to severe immunodeficiency disease in which T lymphocytes and B lymphocytes do not develop properly. Lack of ADA leads to a 100-fold increase in the cellular concentration of dATP, a strong inhibitor of ribonucleotide reductase (Fig. 22-44). High levels of dATP produce a general deficiency of other dNTPs in T lymphocytes. The basis for B-lymphocyte toxicity is less clear. Individuals with ADA deficiency lack an effective immune system and do not survive unless treated. Current therapies include bone marrow transplants from a matched donor to replace the hematopoietic stem cells that mature into B and T lymphocytes. However, transplant recipients often suffer a variety of cognitive and physiological problems. Enzyme replacement therapy, requiring once- or twice-weekly intramuscular injection of active ADA, is effective, but the therapeutic benefit often declines after 8 to 10 years; complications may then arise, including malignancies. For many people, a permanent cure requires replacing the defective gene with a functional one in bone marrow cells. ADA deficiency was one of the first targets of human gene therapy trials (in 1990). Mixed results in early trials have given way to significant successes, and gene therapy is rapidly becoming a viable path for long-term restoration of immune function for these patients. Newer approaches based on CRISPR-mediated gene editing (see Fig. 9-21) may eventually be even more effective.
Purine and Pyrimidine Bases Are Recycled by Salvage Pathways
Free purine and pyrimidine bases are constantly released in cells during the metabolic degradation of nucleotides. Free purines are in large part salvaged and reused to make nucleotides, in a pathway much simpler than the de novo synthesis of purine nucleotides described earlier. One of the primary salvage pathways consists of a single reaction catalyzed by adenosine phosphoribosyltransferase, in which free adenine reacts with PRPP to yield the corresponding adenine nucleotide:
Free guanine and hypoxanthine (the deamination product of adenine; Fig. 22-48) are salvaged in the same way by hypoxanthine-guanine phosphoribosyltransferase. A similar salvage pathway exists for pyrimidine bases in microorganisms, and possibly in mammals.
A genetic lack of hypoxanthine-guanine phosphoribosyltransferase activity, seen almost exclusively in young boys, results in a set of symptoms called Lesch-Nyhan syndrome. Children with this genetic disorder, which becomes manifest by the age of 2 years, are sometimes poorly coordinated and have intellectual deficits. In addition, they are extremely hostile and show compulsive self-destructive tendencies: they mutilate themselves by biting off their fingers, toes, and lips.
The devastating effects of Lesch-Nyhan syndrome illustrate the importance of the salvage pathways. Hypoxanthine and guanine arise constantly from the breakdown of nucleic acids. In the absence of hypoxanthine-guanine phosphoribosyltransferase, PRPP levels rise and purines are overproduced by the de novo pathway, resulting in high levels of uric acid production and goutlike damage to tissue (see below). The brain is especially dependent on the salvage pathways, and this may account for the central nervous system damage in children with Lesch-Nyhan syndrome. This syndrome is another potential target for gene therapy.
Excess Uric Acid Causes Gout
Long thought (erroneously) to be due to “high living,” gout is a disease of the joints caused by an elevated concentration of uric acid in the blood and tissues. The joints become inflamed, painful, and arthritic, owing to the abnormal deposition of sodium urate crystals. The kidneys are also affected, as excess uric acid is deposited in the kidney tubules. Gout occurs predominantly in males. Its precise cause is not known, but it often involves an underexcretion of urate. A genetic deficiency of one or another enzyme of purine metabolism may also be a factor in some cases.
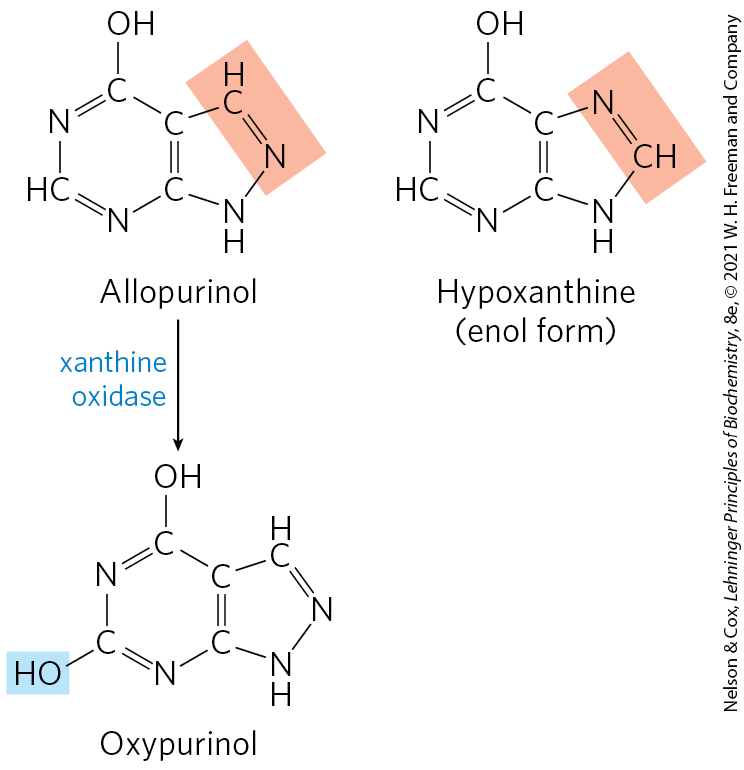
Gout is effectively treated by a combination of nutritional and drug therapies. Patients exclude foods especially rich in nucleotides and nucleic acids, such as liver or glandular products, from the diet. Major alleviation of the symptoms is provided by the drug allopurinol (Fig. 22-50), which inhibits xanthine oxidase, the enzyme that catalyzes the conversion of purines to uric acid. Allopurinol is a substrate of xanthine oxidase, which converts allopurinol to oxypurinol (alloxanthine). Oxypurinol inactivates the reduced form of the enzyme by remaining tightly bound in its active site. When xanthine oxidase is inhibited, the excreted products of purine metabolism are xanthine and hypoxanthine, which are more water-soluble than uric acid and less likely to form crystalline deposits. Allopurinol was developed by Gertrude Elion and George Hitchings, who also developed acyclovir, used in treating people with genital and oral herpes infections, and other purine analogs used in cancer chemotherapy.
FIGURE 22-50 Allopurinol, an inhibitor of xanthine oxidase. Hypoxanthine is the normal substrate of xanthine oxidase. A slight alteration in the structure of hypoxanthine (shaded light red) yields the medically effective enzyme inhibitor allopurinol. At the active site, allopurinol is converted to oxypurinol, a strong competitive inhibitor that remains tightly bound to the reduced form of the enzyme.
George Hitchings, 1905–1998, and Gertrude Elion, 1918–1999.
Many Chemotherapeutic Agents Target Enzymes in Nucleotide Biosynthetic Pathways
The growth of cancer cells is not controlled in the same way as cell growth in most normal tissues. Cancer cells have greater requirements for nucleotides as precursors of DNA and RNA, and consequently are generally more sensitive than normal cells to inhibitors of nucleotide biosynthesis. A number of important chemotherapeutic agents — for cancer and other diseases — act by inhibiting one or more enzymes in these pathways. Several well-studied examples can illustrate productive approaches to treatment and help us understand how these enzymes work.
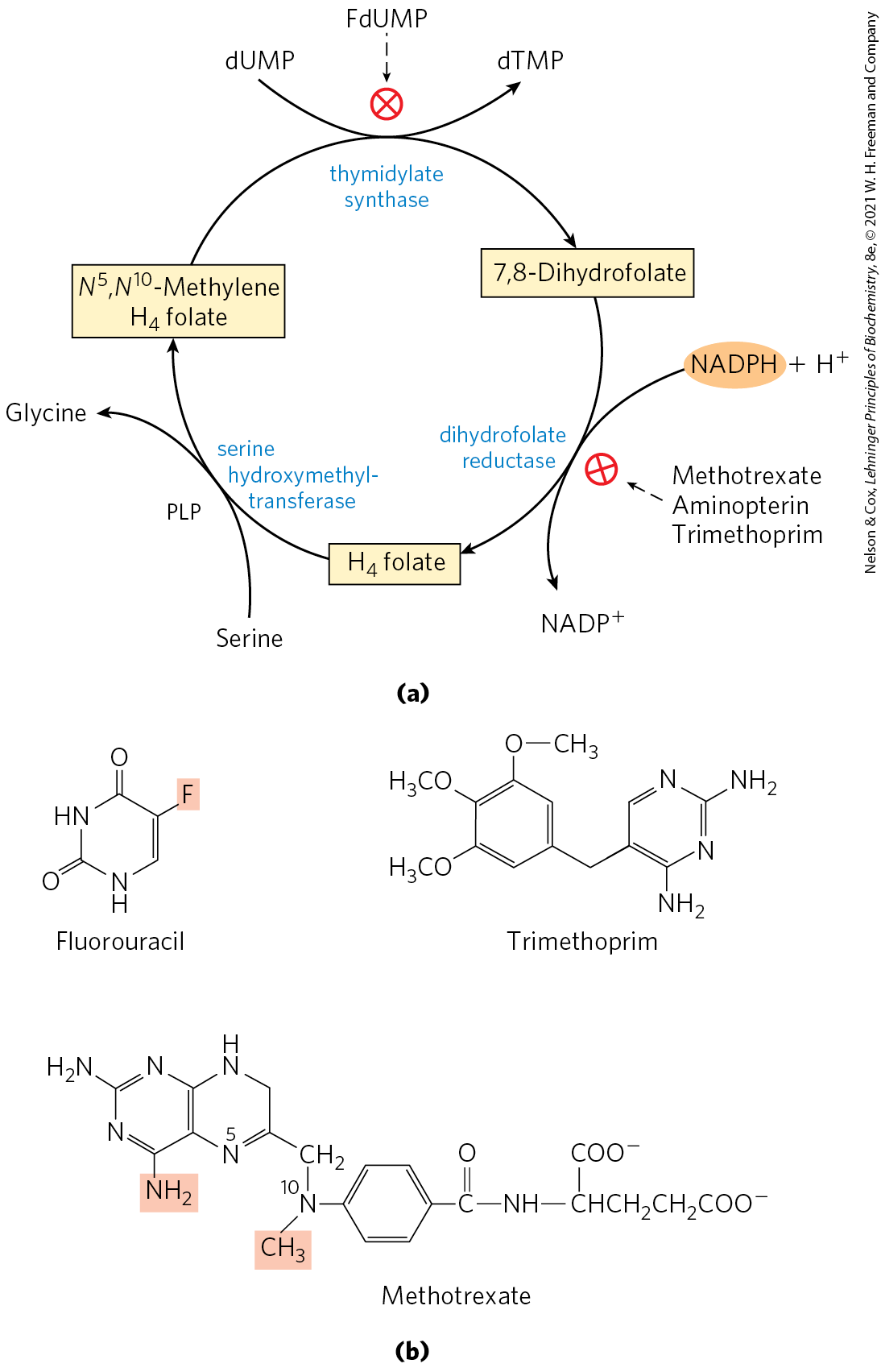
Important targets for pharmaceutical agents include thymidylate synthase and dihydrofolate reductase, enzymes that provide the only cellular pathway for thymine synthesis (Fig. 22-51). One inhibitor that acts on thymidylate synthase, fluorouracil, is an important chemotherapeutic agent. Fluorouracil itself is not the enzyme inhibitor. In the cell, salvage pathways convert it to the deoxynucleoside monophosphate FdUMP, which binds to and inactivates the enzyme. Inhibition by FdUMP (Fig. 22-52) is a classic example of mechanism-based enzyme inactivation. Another prominent chemotherapeutic agent, methotrexate, is an inhibitor of dihydrofolate reductase. This folate analog acts as a competitive inhibitor; the enzyme binds methotrexate with about 100 times higher affinity than dihydrofolate. Aminopterin is a related compound that acts similarly.
FIGURE 22-51 Thymidylate synthesis and folate metabolism as targets of chemotherapy. (a) During thymidylate synthesis, -methylenetetrahydrofolate is converted to 7,8-dihydrofolate; the -methylenetetrahydrofolate is regenerated in two steps (see Fig. 22-47). This cycle is a major target of several chemotherapeutic agents. (b) Fluorouracil and methotrexate are important chemotherapeutic agents. In cells, fluorouracil is converted to FdUMP, which inhibits thymidylate synthase. Methotrexate, a structural analog of tetrahydrofolate, inhibits dihydrofolate reductase; the shaded amino and methyl groups replace a carbonyl oxygen and a proton, respectively, in folate. Another important folate analog, aminopterin, is identical to methotrexate except that it lacks the shaded methyl group. Trimethoprim, a tight-binding inhibitor of bacterial dihydrofolate reductase, was developed as an antibiotic.
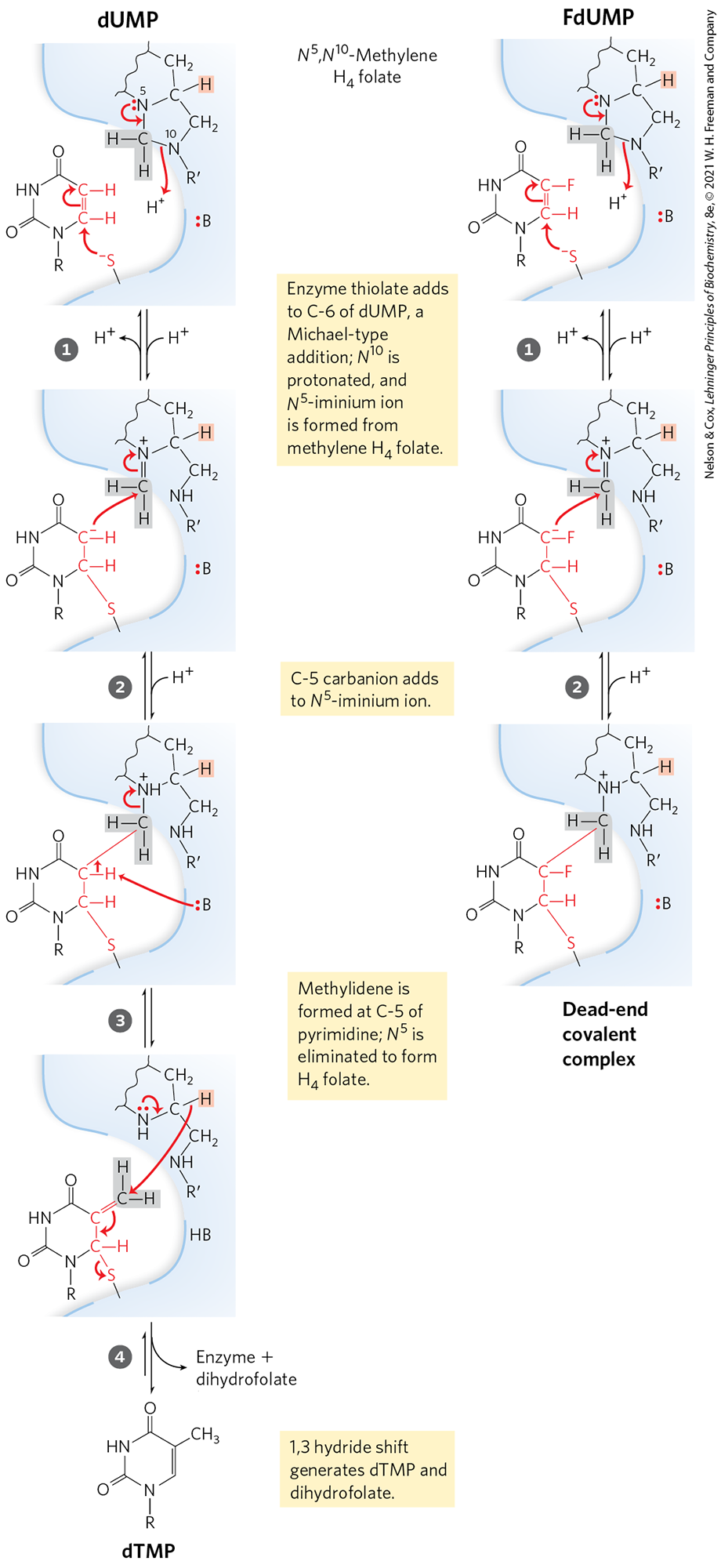
MECHANISM FIGURE 22-52 Conversion of dUMP to dTMP and its inhibition by FdUMP. The normal reaction mechanism of thymidylate synthase (left). The nucleophilic sulfhydryl group contributed by the enzyme in step and the ring atoms of dUMP taking part in the reaction are shown in red; :B denotes an amino acid side chain that acts as a base to abstract a proton after step . The hydrogens derived from the methylene group of -methylenetetrahydrofolate are shaded in gray. The 1,3 hydride shift (step ) moves a hydride ion (shaded light red) from C-6 of tetrahydrofolate to the methyl group of thymidine, oxidizing tetrahydrofolate to dihydrofolate. This hydride shift is blocked when FdUMP is the substrate (right). Steps and proceed normally, but they result in a stable complex — consisting of FdUMP linked covalently to the enzyme and to tetrahydrofolate — that inactivates the enzyme.
The medical potential of inhibitors of nucleotide biosynthesis is not limited to cancer treatment. All fast-growing cells (including bacteria and protists) are potential targets. Trimethoprim, an antibiotic developed by Hitchings and Elion, binds to bacterial dihydrofolate reductase nearly 100,000 times better than to the mammalian enzyme. It is used to treat certain urinary and middle-ear bacterial infections. Parasitic protists, such as the trypanosomes that cause African sleeping sickness (African trypanosomiasis), lack pathways for de novo nucleotide biosynthesis and are particularly sensitive to agents that interfere with their ability to use salvage pathways to scavenge nucleotides from the surrounding environment. Allopurinol (Fig. 22-50) and several similar purine analogs have shown promise for the treatment of African trypanosomiasis and related afflictions. See Box 6-1 for another approach to combating African trypanosomiasis, made possible by advances in our understanding of metabolism and enzyme mechanisms.
SUMMARY 22.4 Biosynthesis and Degradation of Nucleotides
The purine ring system is built up step by step, beginning with 5-phosphoribosylamine. The amino acids glutamine, glycine, and aspartate furnish all the nitrogen atoms of purines. Two ring-closure steps form the purine nucleus.
Purine biosynthesis is regulated by an elaborate system of feedback inhibition.
Pyrimidines are synthesized from carbamoyl phosphate and aspartate, and ribose 5-phosphate is then attached to yield the pyrimidine ribonucleotides.
Pyrimidine biosynthesis is regulated by feedback inhibition of aspartate transcarbamoylase.
Nucleoside monophosphates are converted to their triphosphates by enzymatic phosphorylation reactions.
Ribonucleotides are converted to deoxyribonucleotides by ribonucleotide reductase, an enzyme with novel mechanistic and regulatory characteristics.
The thymine nucleotides are derived from dCDP and dUMP.
Uric acid and urea are the end products of purine and pyrimidine degradation.
Free purines can be salvaged and rebuilt into nucleotides. Genetic deficiencies in certain salvage enzymes cause serious disorders such as Lesch-Nyhan syndrome.
Accumulation of uric acid crystals in the joints, possibly caused by another genetic deficiency, results in gout.
Enzymes of the nucleotide biosynthetic pathways are targets for an array of chemotherapeutic agents used to treat cancer and other diseases.
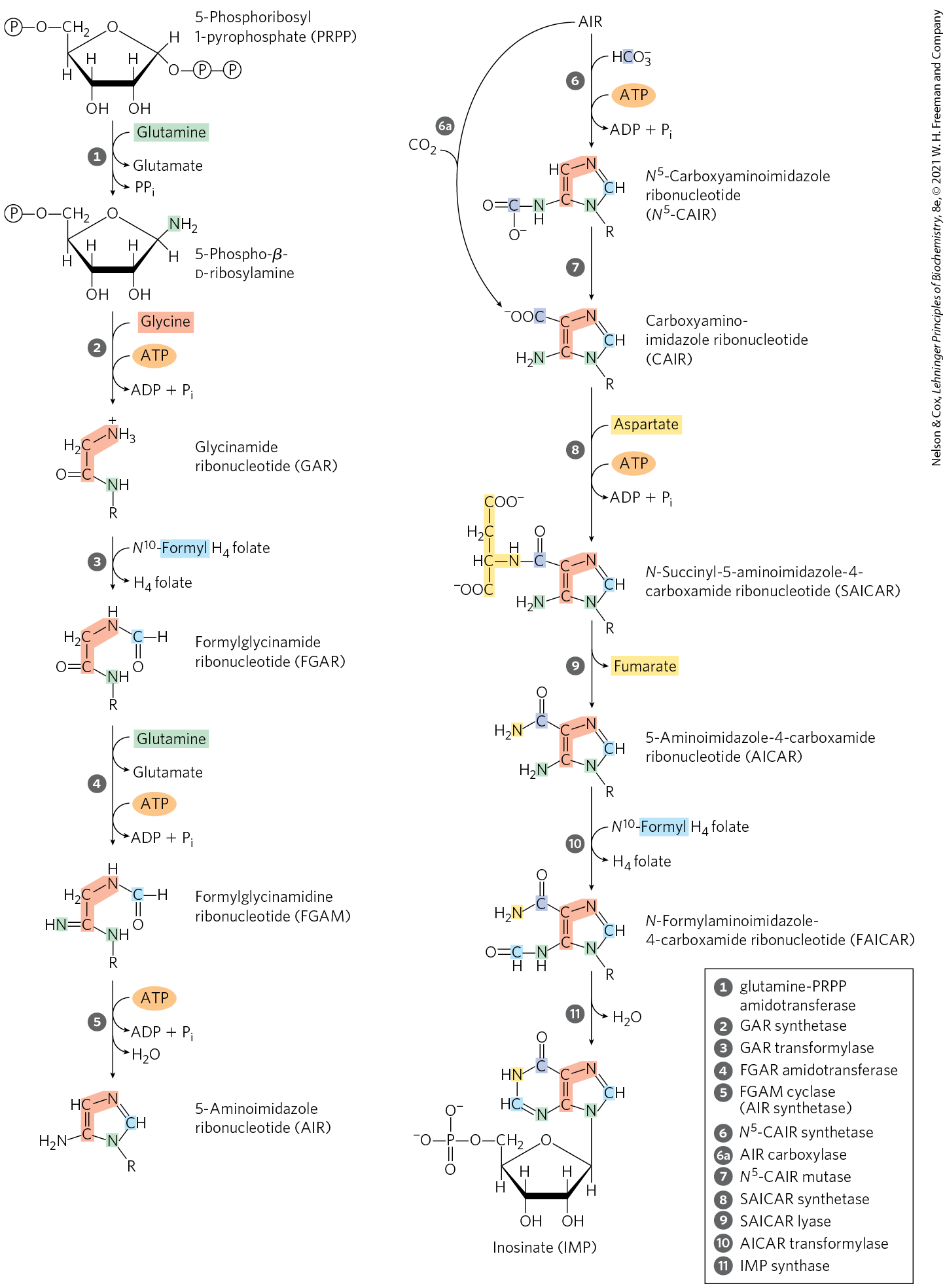
 , R symbolizes the 5-phospho-
, R symbolizes the 5-phospho-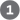 ) is the first committed step in purine synthesis. Note that the product of step
) is the first committed step in purine synthesis. Note that the product of step  , AICAR, is the remnant of ATP released during histidine biosynthesis (see
, AICAR, is the remnant of ATP released during histidine biosynthesis (see  ). Abbreviations are given for most intermediates to simplify the naming of the enzymes. Step
). Abbreviations are given for most intermediates to simplify the naming of the enzymes. Step  is the alternative path from AIR to CAIR occurring in higher eukaryotes.
is the alternative path from AIR to CAIR occurring in higher eukaryotes. ), and a nitrogen is contributed by glutamine (step
), and a nitrogen is contributed by glutamine (step  ), before dehydration and ring closure yield the five-membered imidazole ring of the purine nucleus, as 5-aminoimidazole ribonucleotide (AIR; step
), before dehydration and ring closure yield the five-membered imidazole ring of the purine nucleus, as 5-aminoimidazole ribonucleotide (AIR; step  ). This carboxylation is unusual in that it does not require biotin, but instead uses the bicarbonate generally present in aqueous solutions. A rearrangement transfers the carboxylate from the exocyclic amino group to position 4 of the imidazole ring (step
). This carboxylation is unusual in that it does not require biotin, but instead uses the bicarbonate generally present in aqueous solutions. A rearrangement transfers the carboxylate from the exocyclic amino group to position 4 of the imidazole ring (step  ). Steps
). Steps  and
and  ), and a second ring closure takes place to yield the second fused ring of the purine nucleus (step
), and a second ring closure takes place to yield the second fused ring of the purine nucleus (step  ). The first intermediate with a complete purine ring is inosinate (IMP).
). The first intermediate with a complete purine ring is inosinate (IMP).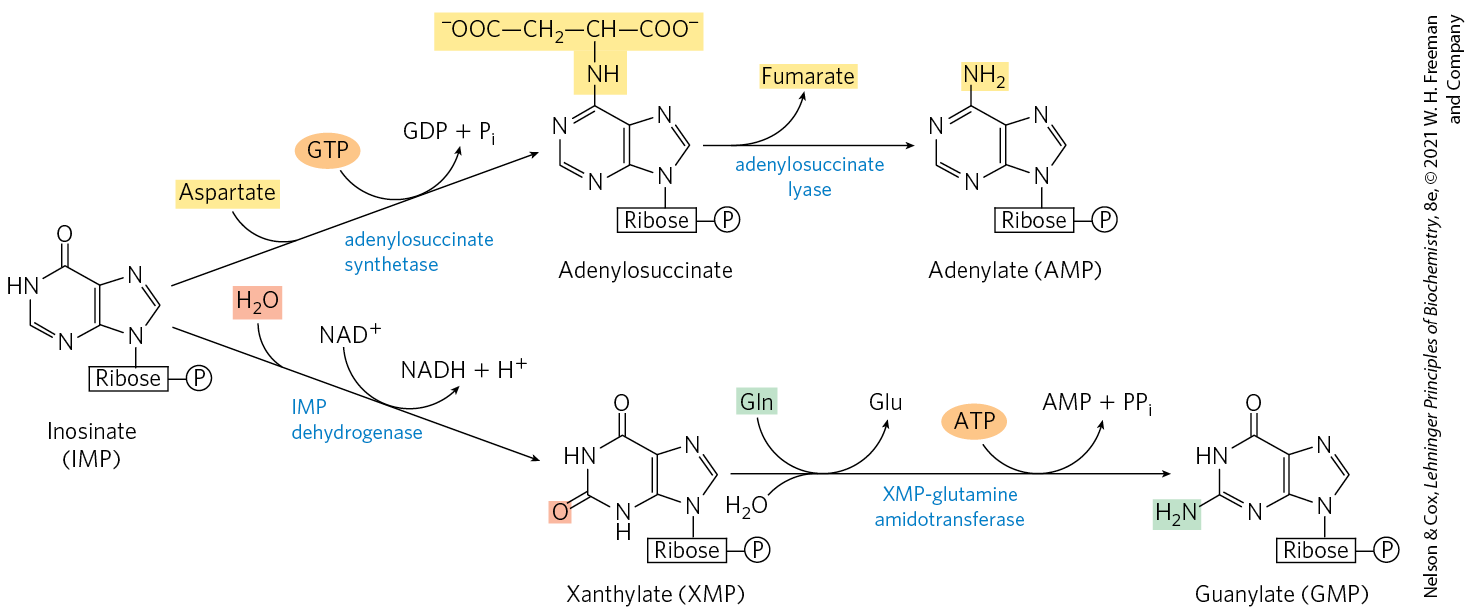
 Four major feedback mechanisms cooperate in regulating the overall rate of de novo purine nucleotide synthesis and the relative rates of formation of the two
Four major feedback mechanisms cooperate in regulating the overall rate of de novo purine nucleotide synthesis and the relative rates of formation of the two 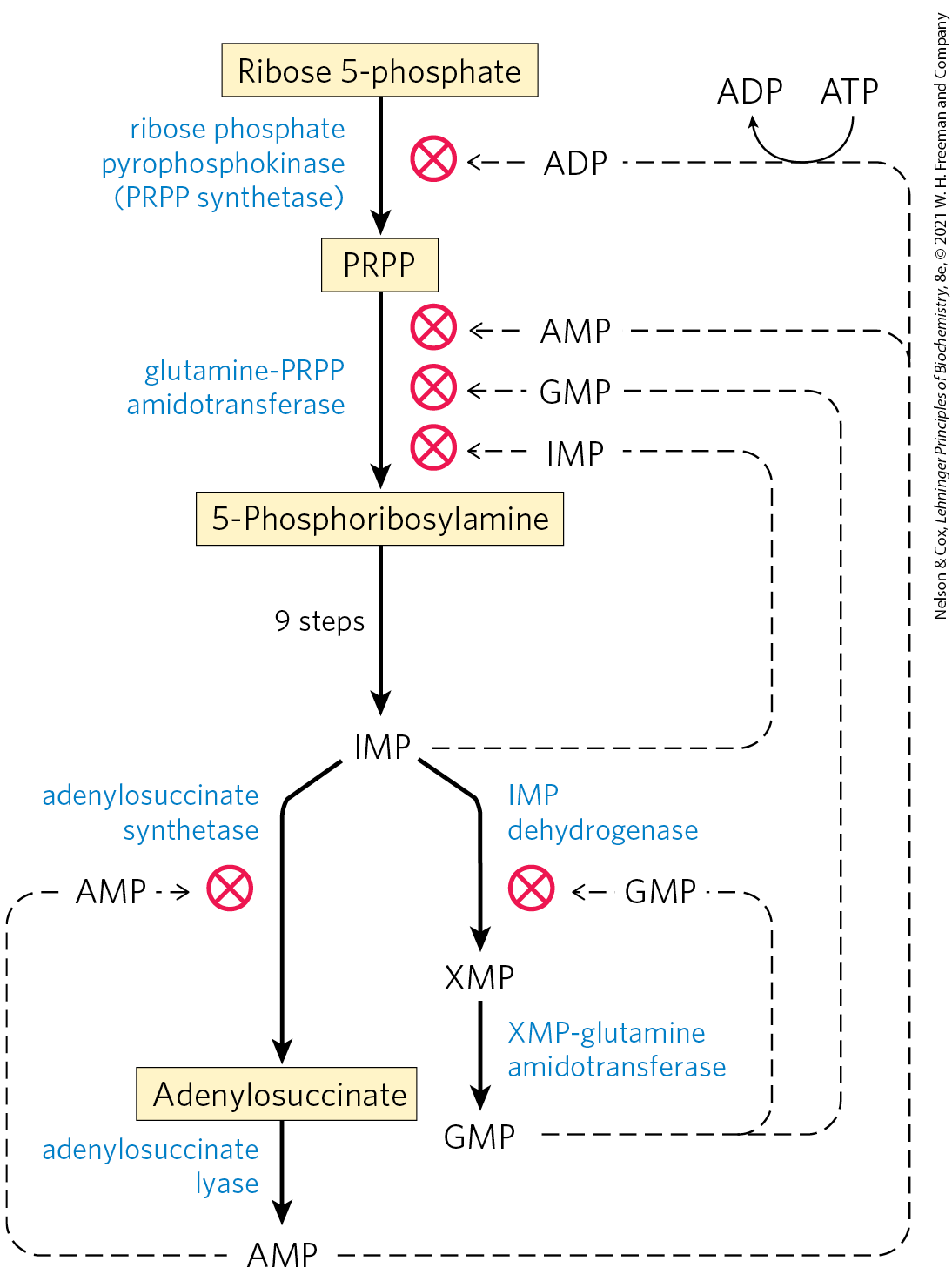
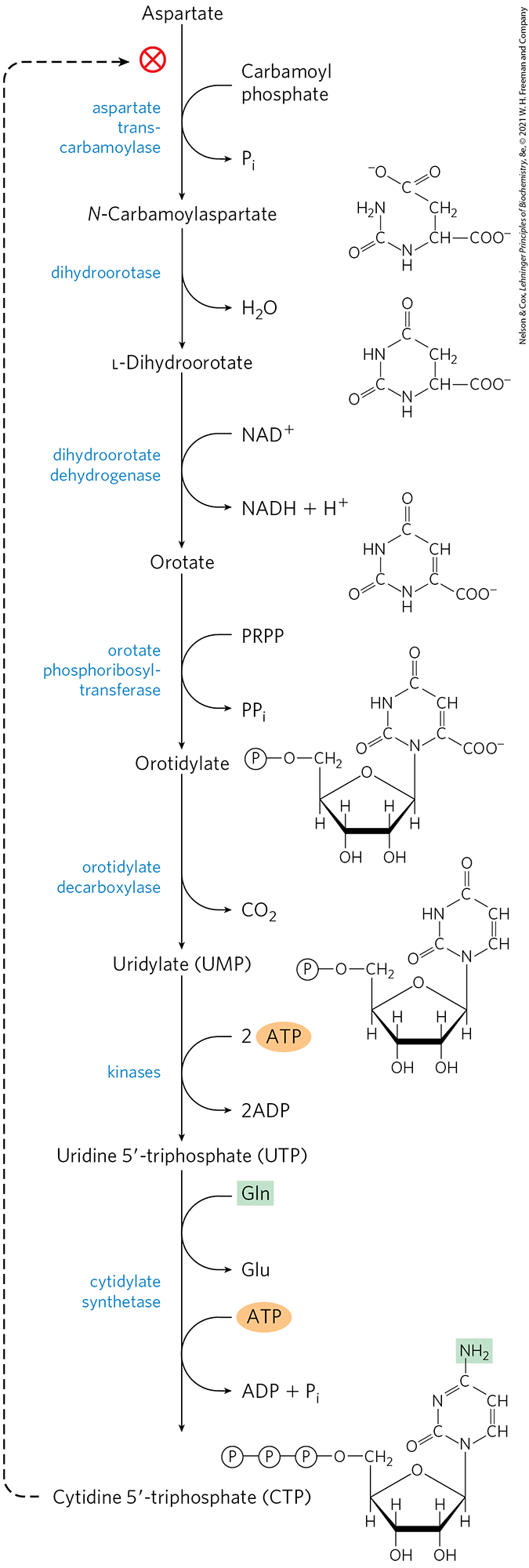
 CTP is formed from UTP by the action of cytidylate synthetase, by way of an acyl phosphate intermediate (consuming one ATP). The nitrogen donor is normally glutamine, although the cytidylate synthetases in many species can use directly.
CTP is formed from UTP by the action of cytidylate synthetase, by way of an acyl phosphate intermediate (consuming one ATP). The nitrogen donor is normally glutamine, although the cytidylate synthetases in many species can use directly.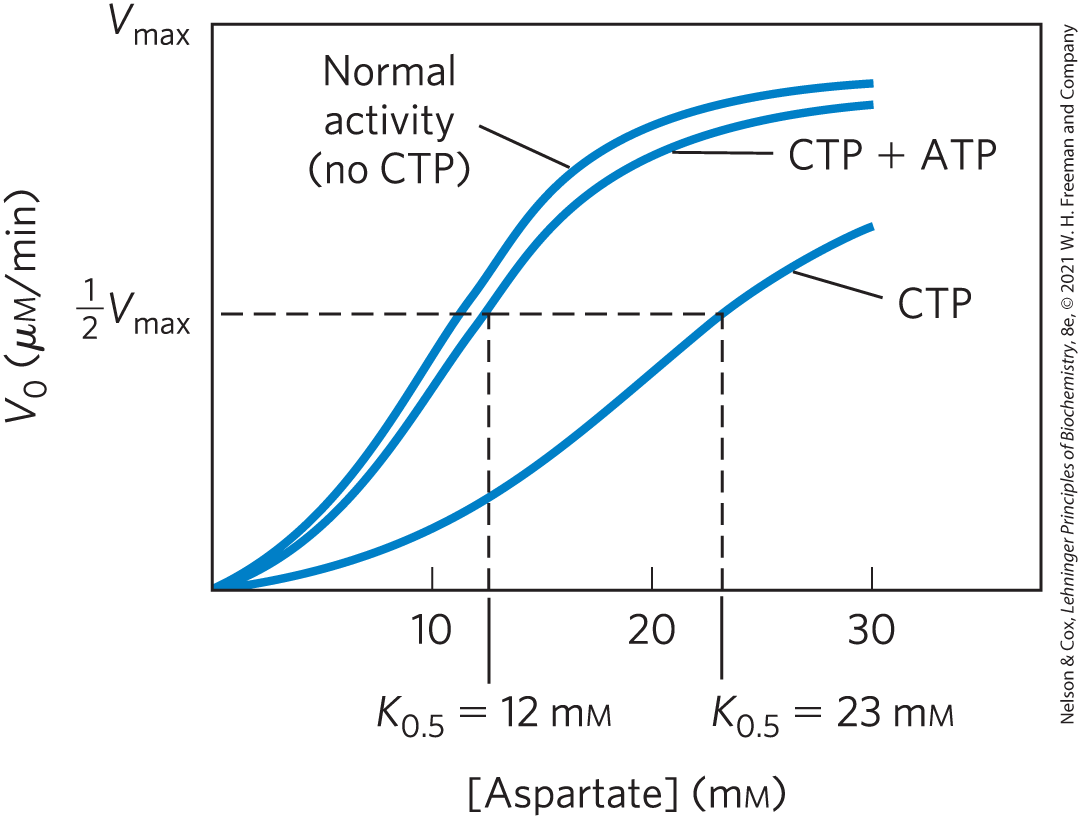

 The scheme is designed to provide a balanced pool of precursors for DNA synthesis. ATP is also a general activator for biosynthesis and ribonucleotide reduction. The presence of dATP in small amounts increases the reduction of pyrimidine nucleotides. An oversupply of the pyrimidine dNTPs is signaled by high levels of dTTP. Abundant dTTP shifts the specificity to favor reduction of GDP. High levels of dGTP, in turn, shift the specificity to ADP reduction, and high levels of dATP shut the enzyme down. These effectors are thought to induce several distinct enzyme conformations with altered specificities.
The scheme is designed to provide a balanced pool of precursors for DNA synthesis. ATP is also a general activator for biosynthesis and ribonucleotide reduction. The presence of dATP in small amounts increases the reduction of pyrimidine nucleotides. An oversupply of the pyrimidine dNTPs is signaled by high levels of dTTP. Abundant dTTP shifts the specificity to favor reduction of GDP. High levels of dGTP, in turn, shift the specificity to ADP reduction, and high levels of dATP shut the enzyme down. These effectors are thought to induce several distinct enzyme conformations with altered specificities.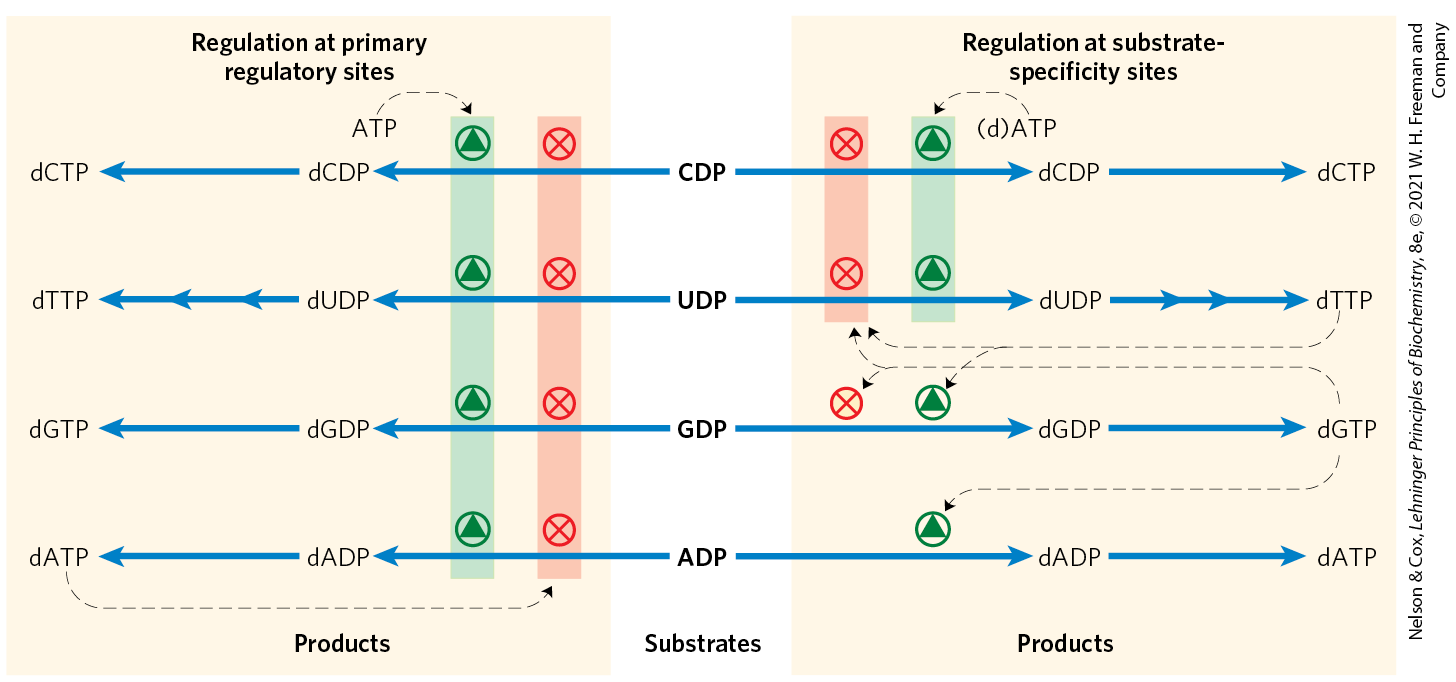
 About 10% of the human population (and up to 50% of people in impoverished communities) suffers from folic acid deficiency. When the deficiency is severe, the symptoms can include heart disease, cancer, and some types of brain dysfunction. Folic acid deficiency during pregnancy can also produce neural tube defects in infants. At least some of these symptoms arise from a reduction in thymidylate synthesis, leading to an abnormal incorporation of uracil into DNA. Uracil is recognized by DNA repair pathways (described in
About 10% of the human population (and up to 50% of people in impoverished communities) suffers from folic acid deficiency. When the deficiency is severe, the symptoms can include heart disease, cancer, and some types of brain dysfunction. Folic acid deficiency during pregnancy can also produce neural tube defects in infants. At least some of these symptoms arise from a reduction in thymidylate synthesis, leading to an abnormal incorporation of uracil into DNA. Uracil is recognized by DNA repair pathways (described in 

 The purine ring system is built up step by step, beginning with 5-phosphoribosylamine. The amino acids glutamine, glycine, and aspartate furnish all the nitrogen atoms of purines. Two ring-closure steps form the purine nucleus.
The purine ring system is built up step by step, beginning with 5-phosphoribosylamine. The amino acids glutamine, glycine, and aspartate furnish all the nitrogen atoms of purines. Two ring-closure steps form the purine nucleus.