9.2 Exploring Protein Function on the Scale of Cells or Whole Organisms
Protein function can be described on three levels. Phenotypic function describes the effects of a protein on the entire organism. For example, loss of the protein may lead to slower growth of the organism, an altered development pattern, or even death. Cellular function is a description of the network of interactions a protein engages in at the cellular level. Identifying interactions with other proteins in the cell can help define the kinds of metabolic processes in which the protein participates. Finally, molecular function refers to the precise biochemical activity of a protein, including details such as the reactions that an enzyme catalyzes or the ligands that a receptor binds. In response to the challenge of understanding these functions of the thousands of proteins in a typical cell, scientists have developed a variety of techniques in the broader discipline of genomics. We can apply these techniques to determine when a particular protein is expressed, what other proteins it might be related to, where it is located in the cell, what other cellular components it interacts with, and what happens to the cell when the protein is missing.
A variety of interrelated methods broadly probe a cell’s RNA or protein content. The entire complement of transcribed RNAs present at a given moment in a cell is defined as the cellular transcriptome. As introduced in Chapters 1 and 3, the entire complement of proteins present at a given moment in a cell is defined as that cell’s proteome. Studies of transcriptomes and studies of proteomes are referred to as transcriptomics and proteomics, respectively. Changes in these cellular macromolecules that occur when a particular gene or its expression is altered can provide important additional clues about protein function, as we will see. The methods we cover here are summarized in Table 9-4. The list is by no means comprehensive, but it serves to illustrate important approaches.
Clue |
Method to Apply |
|---|---|
What is a protein’s function? |
|
What other proteins of known function have similar sequences? |
Comparative genomics |
What known sequence motifs does the protein possess? |
Comparative genomics |
Under what conditions is the gene encoding the protein expressed? |
RNA-Seq |
How much of the protein is present in the cell under different conditions? |
Mass spectrometry |
Where is the protein located in the cell? |
Microscopy with fusion proteins and immunofluorescence |
What does the protein interact with? |
Immunoprecipitation; tandem affinity purification; yeast two-hybrid analysis |
What happens to the cell when the protein is missing or altered? |
CRISPR/Cas9 or other mutagenic methods |
What genes (some unknown) are involved in a process? |
Large-scale screening |
Sequence or Structural Relationships Can Suggest Protein Function
One important reason to sequence many genomes is to provide databases that can be used to assign gene functions by genome comparisons, an enterprise referred to as comparative genomics.
 One important reason to sequence many genomes is to provide databases that can be used to assign gene functions by genome comparisons, an enterprise referred to as
One important reason to sequence many genomes is to provide databases that can be used to assign gene functions by genome comparisons, an enterprise referred to as A genome sequence is simply a very long string of A, G, T, and C residues, all meaningless until interpreted. Genome annotation yields information about the location and function of genes and other critical sequences. Genome annotation converts the sequence into information that any researcher can use, and it is typically focused on genomic DNA encompassing genes that encode RNA and protein, the most common targets of scientific investigation. Every newly sequenced genome includes many genes — often 40% or more of the total — about which little or nothing is known.
Using online tools that apply computational power to comparative genomics, scientists can define gene locations and assign tentative gene functions (where possible) based on similarity to genes previously studied in other genomes. The classic BLAST (Basic Local Alignment Search Tool) algorithm allows a rapid search of all genome databases for sequences related to one that a researcher is exploring, and it is especially valuable for investigating the function of a particular gene. BLAST is one of many resources available at the NCBI (National Center for Biotechnology Information) site (www.ncbi.nlm.nih.gov), sponsored by the National Institutes of Health, and the Ensembl site (www.ensembl.org), cosponsored by the EMBL-EBI (European Molecular Biology Laboratory–European Bioinformatics Institute).
Comparative genomics is made possible by evolutionary biology. Sometimes a newly discovered gene is related by sequence homologies to a previously studied gene in another or the same species, and its function can be entirely or partly defined by that relationship. Genes that occur in different species but have a clear sequence and functional relationship to each other are called orthologs. Genes similarly related to each other within a single species are called paralogs. We introduced these terms in Chapter 3 in the context of proteins. As with proteins, information about the function of a gene in one species can be used to at least tentatively assign function to the orthologous gene found in a second species. The correlation is easiest to make when comparing genomes from relatively closely related species, such as mouse and human, although many clearly orthologous genes have been identified in species as distant as bacteria and humans. Sometimes even the order of genes on a chromosome is conserved over large segments of the genomes of closely related species (Fig. 9-14). Conserved gene order, called synteny, provides additional evidence for an orthologous relationship between genes at identical locations within the related segments.
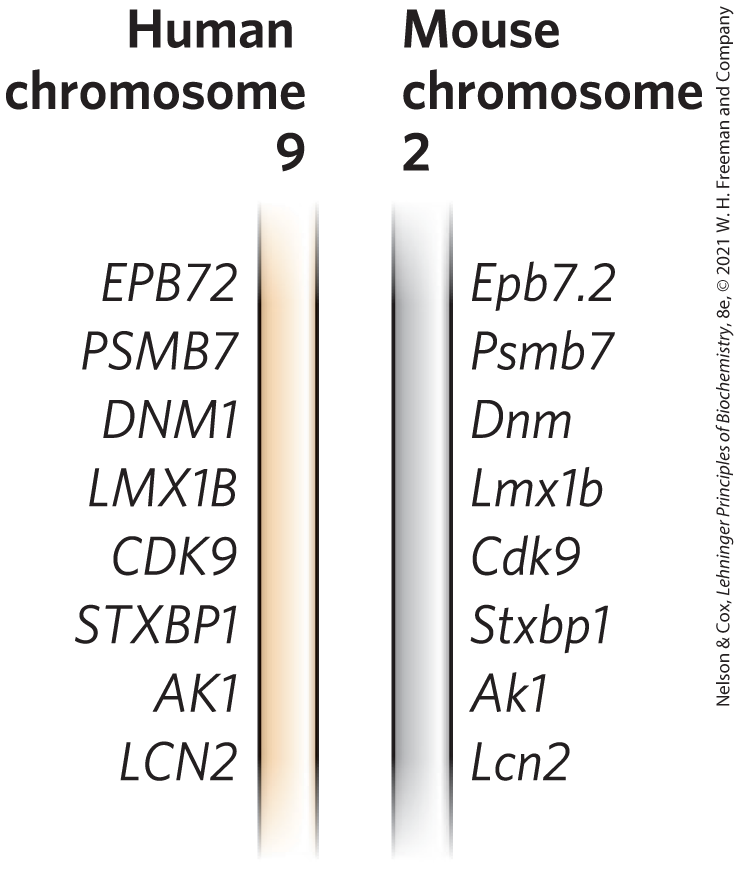
FIGURE 9-14 Synteny in the human and mouse genomes. Large segments of the two genomes have closely related genes aligned in the same order on the chromosomes. In these short segments of human chromosome 9 and mouse chromosome 2, the genes show a very high degree of homology, as well as the same gene order. The different lettering schemes for the gene names simply reflect the different naming conventions for the two species. [Information from T. G. Wolfsberg et al., Nature 409:824, 2001, Fig. 1.]
Alternatively, certain amino acid sequences associated with particular structural motifs (Chapter 4) may be identified within a protein. The presence of a structural motif may help to define molecular function by suggesting that a protein, say, catalyzes ATP hydrolysis, binds to DNA, or forms a complex with zinc ions. These relationships are determined with the aid of sophisticated computer programs, limited only by the current information on gene and protein structure and by our capacity to associate sequences with particular structural motifs. Sequences at an enzyme active site that have been highly conserved during evolution are typically associated with catalytic function, and their identification is often a key step in defining an enzyme’s reaction mechanism. The reaction mechanism, in turn, provides information needed to develop new enzyme inhibitors that can be used as pharmaceutical agents.
 The presence of a structural motif may help to define molecular function by suggesting that a protein, say, catalyzes ATP hydrolysis, binds to DNA, or forms a complex with zinc ions. These relationships are determined with the aid of sophisticated computer programs, limited only by the current information on gene and protein structure and by our capacity to associate sequences with particular structural motifs. Sequences at an enzyme active site that have been highly conserved during evolution are typically associated with catalytic function, and their identification is often a key step in defining an enzyme’s reaction mechanism. The reaction mechanism, in turn, provides information needed to develop new enzyme inhibitors that can be used as pharmaceutical agents.
The presence of a structural motif may help to define molecular function by suggesting that a protein, say, catalyzes ATP hydrolysis, binds to DNA, or forms a complex with zinc ions. These relationships are determined with the aid of sophisticated computer programs, limited only by the current information on gene and protein structure and by our capacity to associate sequences with particular structural motifs. Sequences at an enzyme active site that have been highly conserved during evolution are typically associated with catalytic function, and their identification is often a key step in defining an enzyme’s reaction mechanism. The reaction mechanism, in turn, provides information needed to develop new enzyme inhibitors that can be used as pharmaceutical agents.When and Where a Protein Is Present in a Cell Can Suggest Protein Function
If a protein is involved in a reaction or process, it must be present at the location and at the moment that reaction or process occurs. This aspect of protein function can now be explored at multiple levels and with ever-increasing precision.
RNA-Seq and Transcriptomics
The RNAs that are transcribed from a genome under a given set of conditions can be determined using DNA sequencing methods described in Chapter 8. The approach is called RNA-Seq (Fig. 9-15). RNA is first isolated from a tissue or a population of cells. The RNA is fragmented and converted to double-stranded DNA using reverse transcriptase (see Fig. 9-12a). This DNA is then subjected to deep DNA sequencing, which reveals both the RNAs that are present and the relative abundance of each (if more copies of one RNA are present, they will give rise to more DNA sequencing reads). This method is sensitive enough to apply to single cells, an approach called single cell RNA-Seq, or scRNA-Seq. It allows investigators to catalog the RNAs being transcribed in different parts of a tissue.
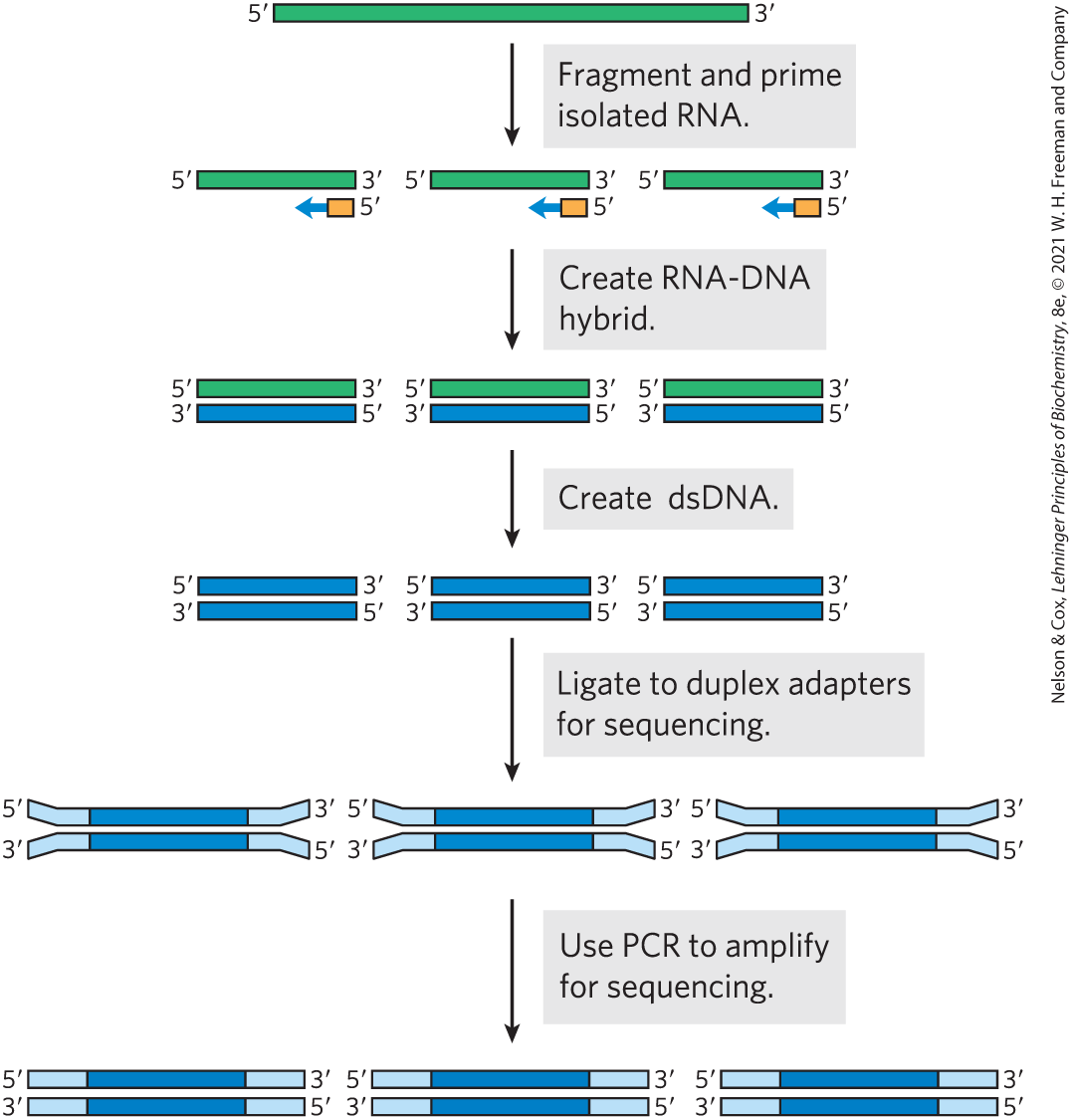
FIGURE 9-15 RNA-Seq. To define a cellular transcriptome, the first step is to isolate cellular RNA. Because many RNAs, particularly mRNAs and rRNAs, are quite long, the RNA is then fragmented to an average size commensurate with the DNA sequencing platform to be used. The RNA is converted to DNA using reverse transcriptase. Hexameric DNA oligonucleotides of random sequence are used to prime the reverse transcriptase if all RNA is to be included in the transcriptome. RNA-DNA hybrids are more stable than DNA-DNA hybrids, so hexameric duplexes are sufficient for this task. If the transcriptome is focused on expression of protein-coding genes, beads coated with poly(dT) can be used to hybridize to the poly(A) tails of eukaryotic mRNAs, allowing their precipitation and enrichment relative to other RNAs. After reverse transcription, the DNA fragments are ligated to duplex adapters that provide a universal priming location for the DNA sequencing as well as sequences that allow annealing to anchors on the sequencing flow cell (see Fig. 8-36). This is followed by deep DNA sequencing and data analysis.
The transcriptional state of a human cell or tissue can be diagnostic of conditions ranging from diabetes to cancer. If a particular gene under study is expressed in a certain tissue or under particular metabolic conditions, the result provides a new functional clue. Detailed knowledge about what genes are expressed in a given tumor may eventually help guide treatment options. RNA-Seq can reveal patterns of gene regulation and expression. It has special importance in studying cancerous tumors, in which rapid evolution triggered by genome instability creates a range of cell types. The mRNAs that are present in tumor cells provide a clue to the proteins that may be present, although not all mRNAs are immediately translated into protein. RNA-Seq also reveals the presence of many types of noncoding RNAs (described in Chapter 26) that are now being defined.
 The transcriptional state of a human cell or tissue can be diagnostic of conditions ranging from diabetes to cancer. If a particular gene under study is expressed in a certain tissue or under particular metabolic conditions, the result provides a new functional clue. Detailed knowledge about what genes are expressed in a given tumor may eventually help guide treatment options. RNA-Seq can reveal patterns of gene regulation and expression. It has special importance in studying cancerous tumors, in which rapid evolution triggered by genome instability creates a range of cell types. The mRNAs that are present in tumor cells provide a clue to the proteins that may be present, although not all mRNAs are immediately translated into protein. RNA-Seq also reveals the presence of many types of noncoding RNAs (described in
The transcriptional state of a human cell or tissue can be diagnostic of conditions ranging from diabetes to cancer. If a particular gene under study is expressed in a certain tissue or under particular metabolic conditions, the result provides a new functional clue. Detailed knowledge about what genes are expressed in a given tumor may eventually help guide treatment options. RNA-Seq can reveal patterns of gene regulation and expression. It has special importance in studying cancerous tumors, in which rapid evolution triggered by genome instability creates a range of cell types. The mRNAs that are present in tumor cells provide a clue to the proteins that may be present, although not all mRNAs are immediately translated into protein. RNA-Seq also reveals the presence of many types of noncoding RNAs (described in 
Cellular Proteomes and Mass Spectrometry
A more direct way to establish protein presence or absence is to assess the cellular proteome. Mass spectrometry (Chapter 3) can accurately catalog and quantify the thousands of proteins present in a typical cell. This approach is a complement to RNA-Seq, as it provides a comprehensive list of the genes that are both transcribed and translated into protein. Mass spectrometry also provides information about how those proteins are modified, in turn allowing an assessment of their regulatory state.
Fusion Proteins and Immunofluorescence
Often, an important clue to the function of a gene product comes from determining its location within the cell. For example, a protein found exclusively in the nucleus could be involved in processes that are unique to that organelle, such as transcription, replication, or chromatin condensation. Researchers often engineer fusion proteins for the purpose of locating a protein in the cell or organism. Some of the most useful fusions are the attachment of marker proteins that signal the location by direct visualization or by immunofluorescence.
 Often, an important clue to the function of a gene product comes from determining its location within the cell. For example, a protein found exclusively in the nucleus could be involved in processes that are unique to that organelle, such as transcription, replication, or chromatin condensation. Researchers often engineer fusion proteins for the purpose of locating a protein in the cell or organism. Some of the most useful fusions are the attachment of marker proteins that signal the location by direct visualization or by immunofluorescence.
Often, an important clue to the function of a gene product comes from determining its location within the cell. For example, a protein found exclusively in the nucleus could be involved in processes that are unique to that organelle, such as transcription, replication, or chromatin condensation. Researchers often engineer fusion proteins for the purpose of locating a protein in the cell or organism. Some of the most useful fusions are the attachment of marker proteins that signal the location by direct visualization or by immunofluorescence.A particularly useful marker is the green fluorescent protein (GFP) (Fig. 9-16), discovered by Osamu Shimomura. As subsequently shown by Martin Chalfie, a target gene (encoding the protein of interest) fused to the GFP gene generates a fusion protein that is highly fluorescent — it literally lights up when exposed to blue light — and can be visualized directly in a living cell. GFP is a protein derived from the jellyfish Aequorea victoria (Fig. 9-16a). The protein has a β-barrel structure with a fluorophore (the fluorescent component of the protein) in the center (Fig. 9-16b). The fluorophore is derived from a rearrangement and oxidation of three amino acid residues (Fig. 9-16c). Because this reaction is autocatalytic and requires no proteins or cofactors other than molecular oxygen, GFP is readily cloned in an active form in almost any cell. Just a few molecules of this protein can be observed microscopically, allowing the study of its location and movements in a cell.
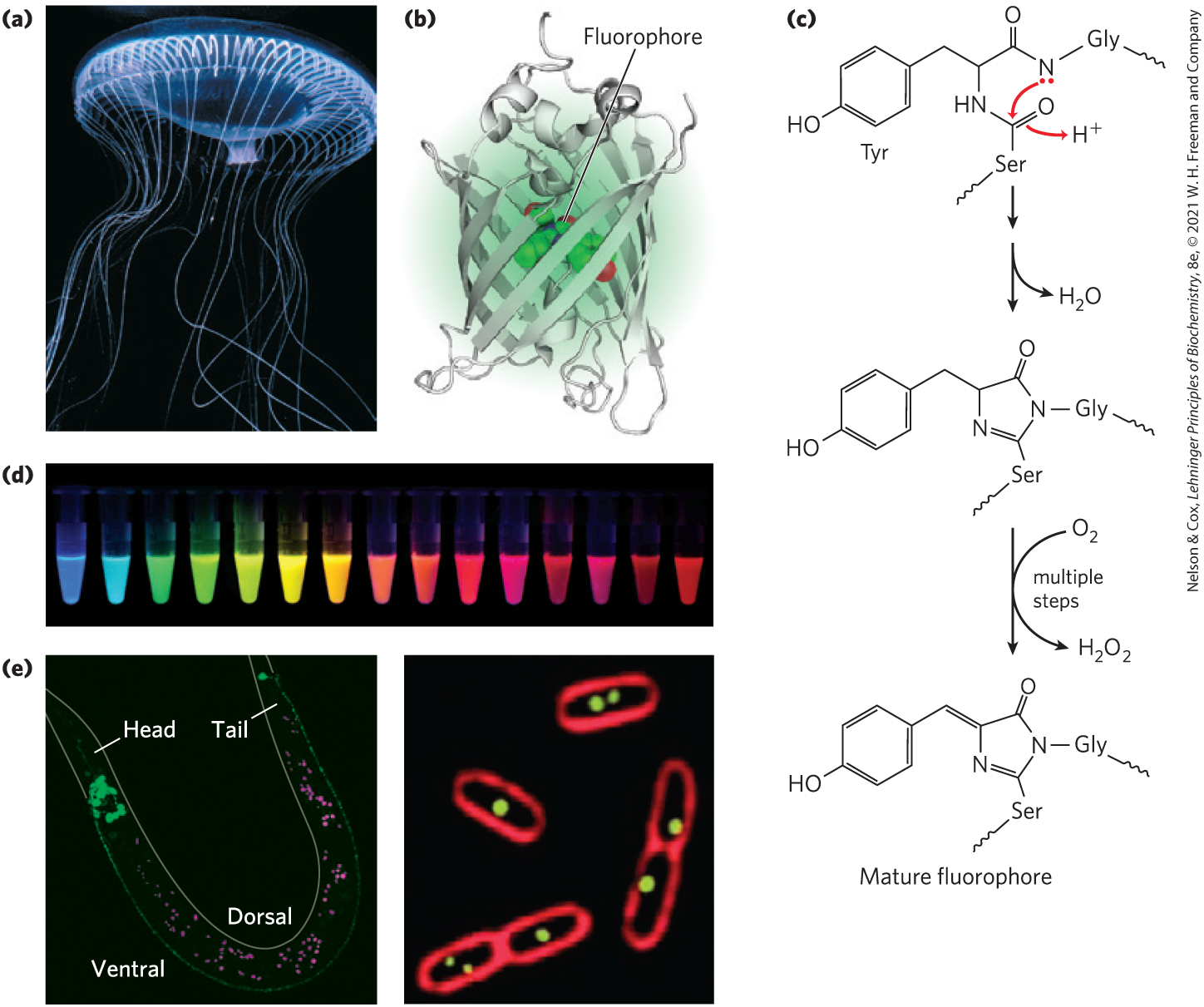
FIGURE 9-16 Green fluorescent protein (GFP). (a) GFP is derived from the jellyfish Aequorea victoria. (b) The protein has a β-barrel structure; the fluorophore is in the center of the barrel. (c) The fluorophore in GFP is derived from a sequence of three amino acids: . The fluorophore achieves its mature form through an internal rearrangement, coupled to a multistep oxidation reaction. An abbreviated mechanism is shown here. (d) Variants of GFP are now available in almost any color of the visible spectrum. (e) A GLR1-GFP fusion protein fluoresces bright green in C. elegans, a nematode worm (left). GLR1 is a glutamate receptor of nervous tissue. (In this photograph, autofluorescing fat droplets are false-colored in magenta.) The membranes of E. coli cells (right) are stained with a red fluorescent dye. The cells are expressing a protein that binds to a resident plasmid, fused to GFP. The green spots indicate the locations of plasmids. [(a) Chris Parks/ImageQuest Marine. (b) Data from PDB ID 1GFL, F. Yang et al., Nature Biotechnol. 14:1246, 1996. (c) Information from Roger Tsien, University of California, San Francisco, Department of Pharmacology, and Paul Steinbach. (d) Courtesy of Roger Tsien and Paul Steinbach, University of California, San Diego, Department of Pharmacology. (e) (left) Courtesy Penelope J. Brockie and Andres V. Maricq, Department of Biology, University of Utah; (right) Courtesy Joseph A. Pogliano, from J. Pogliano et al. (2001), Multicopy plasmids are clustered and localized in Escherichia coli, Proc. Natl. Acad. Sci. USA 98:4486–4491.]
Careful protein engineering by Roger Tsien, coupled with the isolation of related fluorescent proteins from other marine coelenterates, has made variants of these proteins available in an array of colors (Fig. 9-16d) and other characteristics (brightness, stability). If fusion to GFP does not impair the function or properties of a protein one wishes to study, the fusion protein can be used to reveal the protein’s location in the cell under a range of conditions and to detect interactions with other labeled proteins. With this technology, for example, the protein GLR1 (a glutamate receptor of nervous tissue) has been visualized as a GLR1-GFP fusion protein in the nematode Caenorhabditis elegans (Fig. 9-16e).
In some cases, the GFP fusion protein may be inactive or may not be expressed at sufficient levels to allow visualization. Immunofluorescence is an alternative approach for visualizing the endogenous (unaltered) protein. This approach requires fixation (and thus death) of the cell. The protein of interest is sometimes expressed as a fusion protein with an epitope tag, a short protein sequence that is bound tightly by a well-characterized, commercially available antibody. Fluorescent molecules (fluorochromes) are attached to this antibody. More commonly, the target protein is unaltered and is bound by an antibody that is specific for the protein. Next, a second antibody is added that binds specifically to the first one, and it is the second antibody that has the attached fluorochrome(s) (Fig. 9-17). A variation of this indirect approach to visualization is to attach biotin molecules to the first antibody, then add streptavidin (a bacterial protein closely related to avidin, a protein that binds biotin; see Table 5-1) complexed with fluorochromes. The interaction between biotin and streptavidin is one of the strongest and most specific known, and the potential to add multiple fluorochromes to each target protein gives this method great sensitivity. In all of these cases, the end product is a microscopic view of a cell in which a spot of light (a focus) reveals the location of the protein.
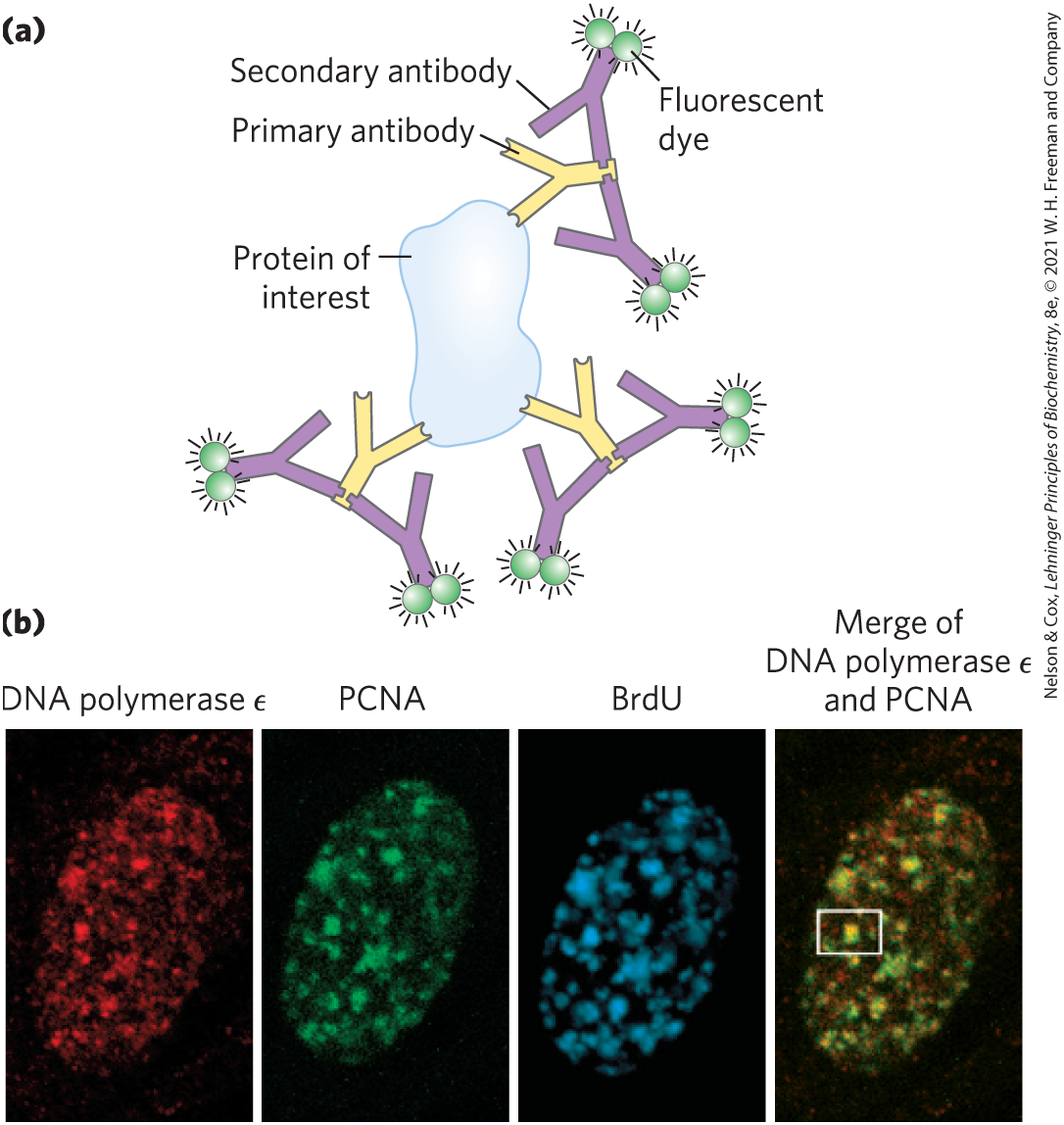
FIGURE 9-17 Indirect immunofluorescence. (a) The protein of interest is bound to a primary antibody, and a secondary antibody is added; this second antibody, with one or more attached fluorescent groups, binds to the first. Multiple secondary antibodies can bind the primary antibody, amplifying the signal. If the protein of interest is in the interior of the cell, the cell is fixed and permeabilized, and the two antibodies are added in succession. (b) The end result is an image in which bright spots indicate the location of the protein or proteins of interest in the cell. The images here show a nucleus from a human fibroblast, successively stained with antibodies and fluorescent labels for DNA polymerase ɛ; for PCNA, an important polymerase accessory protein; and for bromo-deoxyuridine (BrdU), a nucleotide analog. The BrdU, added as a brief pulse, identifies regions undergoing active DNA replication. The patterns of staining show that DNA polymerase ɛ and PCNA co-localize to regions of active DNA synthesis (rightmost image); one such region is visible in the white box. [(b) Fuss, J. and Linn, S., 2002, “Human DNA Polymerase ε Colocalizes with Proliferating Cell Nuclear Antigen and DNA Replication Late, but Not Early, in S Phase,” J. Biol. Chem. 277:8658–8666. Courtesy Jill Fuss, University of California, Berkeley.]
Knowing What a Protein Interacts with Can Suggest Its Function
Another key to defining the function of a particular protein is to determine its biochemical playmates. In the case of protein-protein interactions, the association of a protein of unknown function with one whose function is known can compellingly imply a functional relationship. The techniques used in this effort are quite varied.
Purification of Protein Complexes
By fusing the gene encoding a protein under study with the gene for an epitope tag, investigators can precipitate the protein product of the fusion gene by complexing it with the antibody that binds the epitope. This process is called immunoprecipitation (Fig. 9-18). If the tagged protein is expressed in cells, other proteins that bind to it precipitate with it. Identifying the associated proteins reveals some of the intracellular protein-protein interactions of the tagged protein. There are many variations of this process. For example, a crude extract of cells that express a tagged protein is added to a column containing immobilized antibody (see Fig. 3-17c for a description of affinity chromatography). The tagged protein binds to the antibody, and proteins that interact with the tagged protein are sometimes also retained on the column. The connection between the protein and the tag is cleaved with a specific protease. The protein complexes are eluted from the column, and the proteins in them are identified by mass spectrometry. Researchers can use these methods to define complex networks of interactions within a cell. In principle, the chromatographic approach to analyzing protein-protein interactions can be used with any type of protein tag (His tag, GST, etc.) that can be immobilized on a suitable chromatographic medium.
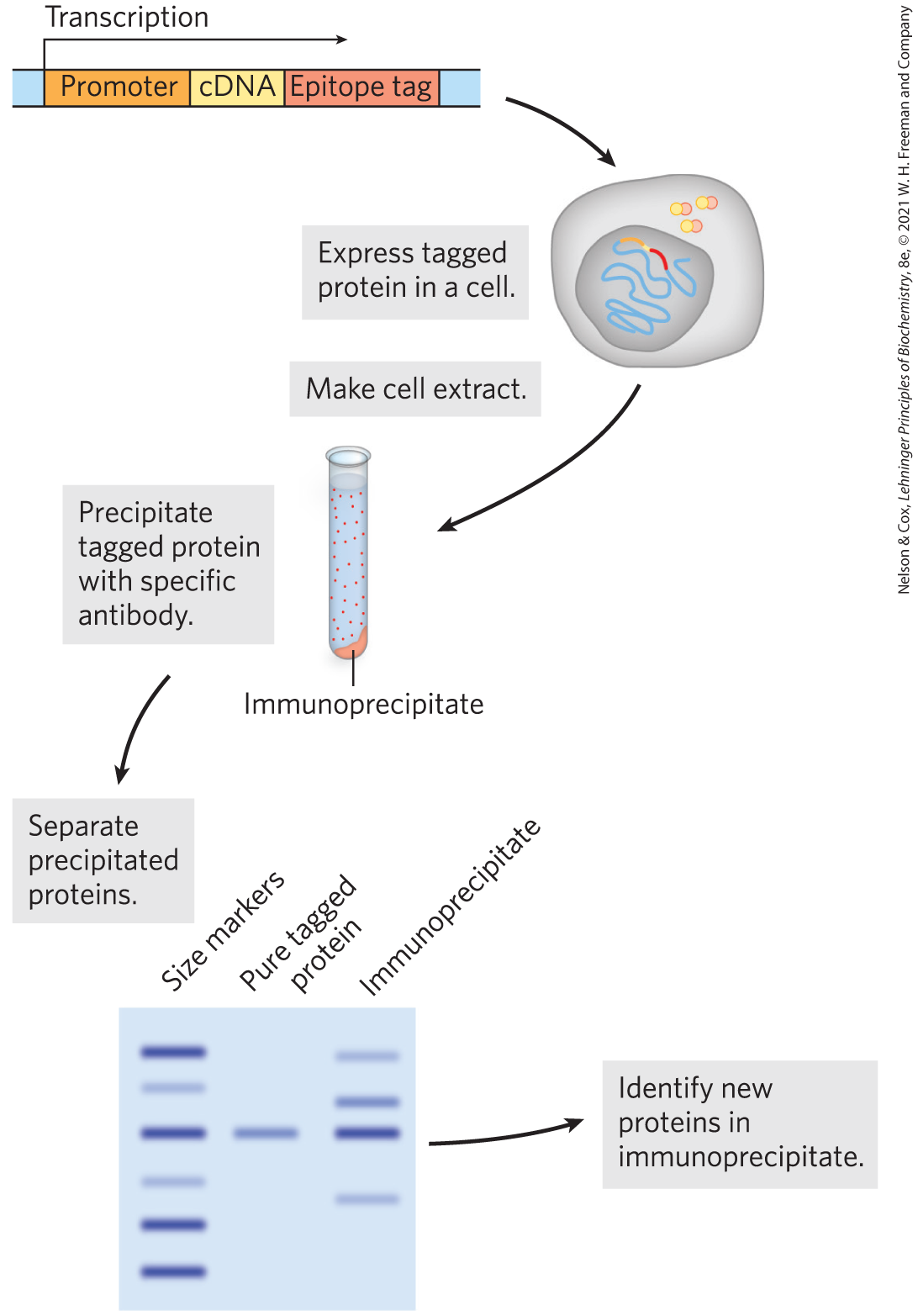
FIGURE 9-18 The use of epitope tags to study protein-protein interactions. The gene of interest is cloned next to a gene for an epitope tag, and the resulting fusion protein is precipitated by antibodies to the epitope. Any other proteins that interact with the tagged protein also precipitate, thereby helping to elucidate protein-protein interactions.
The selectivity of this approach can be enhanced with tandem affinity purification (TAP) tags. Two consecutive tags are fused to a target protein, and the fusion protein is expressed in a cell (Fig. 9-19). The first tag is protein A, a protein found at the surface of the bacterium Staphylococcus aureus that binds tightly to mammalian immunoglobulin G (IgG). The second tag is often a calmodulin-binding peptide. A crude extract containing the TAP-tagged fusion protein is passed through a column matrix with attached IgG antibodies that bind protein A. Most of the unbound cellular proteins are washed through the column, but proteins that normally interact with the target protein in the cell are retained. The first tag is then cleaved from the fusion protein with a highly specific protease, TEV protease, and the shortened fusion target protein and any proteins associated noncovalently with the target protein are eluted from the column. The eluate is then passed through a second column containing a matrix with attached calmodulin that binds the second tag. Loosely bound proteins are again washed from the column. After the second tag is cleaved, the target protein is eluted from the column with its associated proteins. The two consecutive purification steps eliminate most weakly bound contaminants. False positives are minimized, and protein interactions that persist through both steps are likely to be functionally significant.
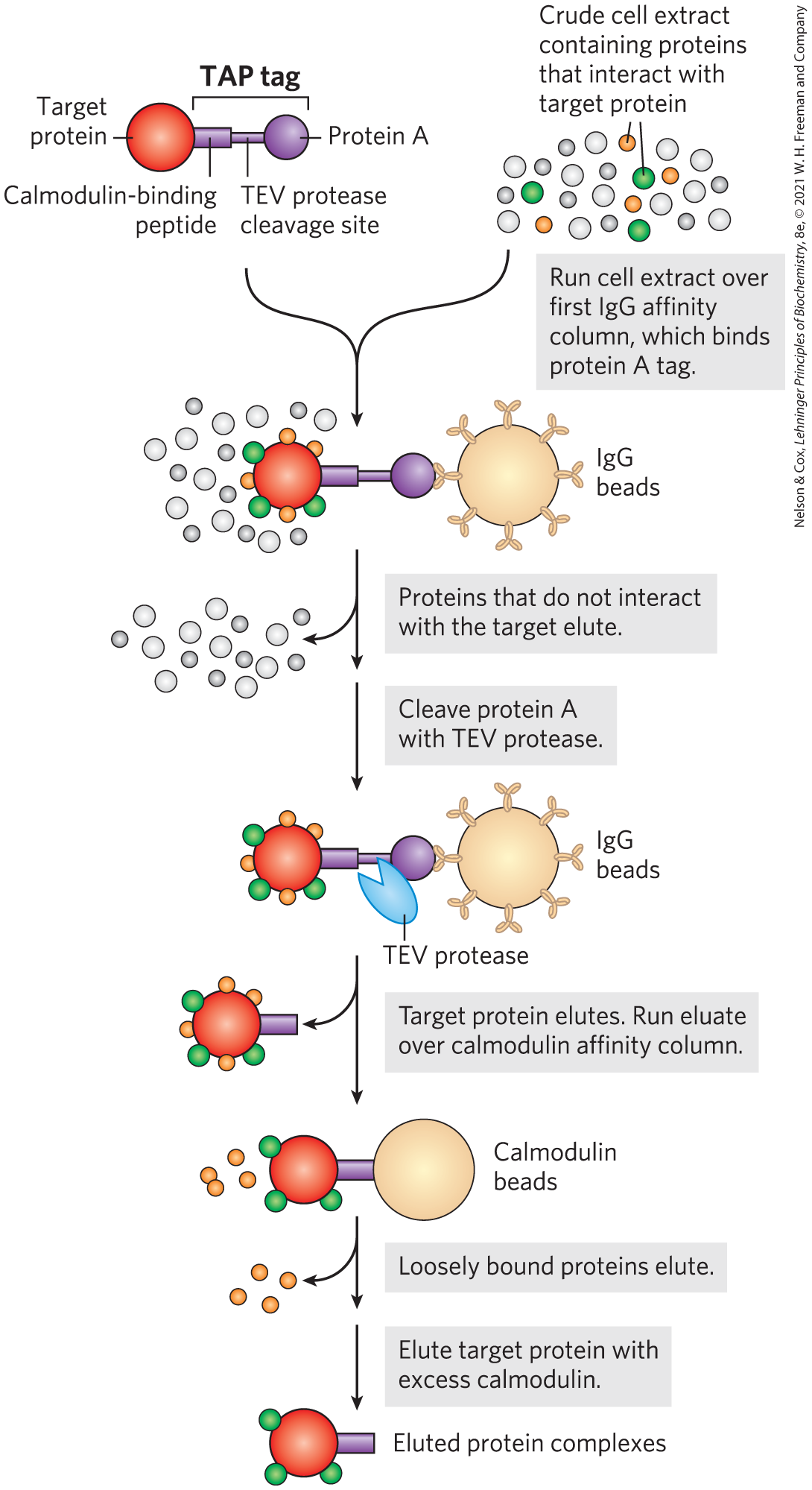
FIGURE 9-19 Tandem affinity purification (TAP) tags. A TAP-tagged protein and associated proteins are isolated by two consecutive affinity purifications, as described in the text.
Yeast Two-Hybrid Analysis
A sophisticated genetic approach to defining protein-protein interactions is based on the properties of the Gal4 protein (Gal4p; see Fig. 28-32), which activates the transcription of GAL genes (encoding the enzymes of galactose metabolism) in yeast. Gal4p has two domains: one that binds a specific DNA sequence, and another that activates RNA polymerase to synthesize mRNA from an adjacent gene. The two domains of Gal4p are stable when separated, but activation of RNA polymerase requires interaction with the activation domain, which in turn requires positioning by the DNA-binding domain. Hence, the domains must be brought together to function correctly.
In yeast two-hybrid analysis, the protein-coding regions of the genes to be analyzed are fused to the yeast gene for either the DNA-binding domain or the activation domain of Gal4p, and the resulting genes express a series of fusion proteins (Fig. 9-20). If a protein fused to the DNA-binding domain interacts with a protein fused to the activation domain, transcription is activated. The reporter gene transcribed by this activation is generally one that yields a protein required for growth or an enzyme that catalyzes a reaction with a colored product. Thus, when grown on the proper medium, cells that contain a pair of interacting proteins are easily distinguished from those that do not.
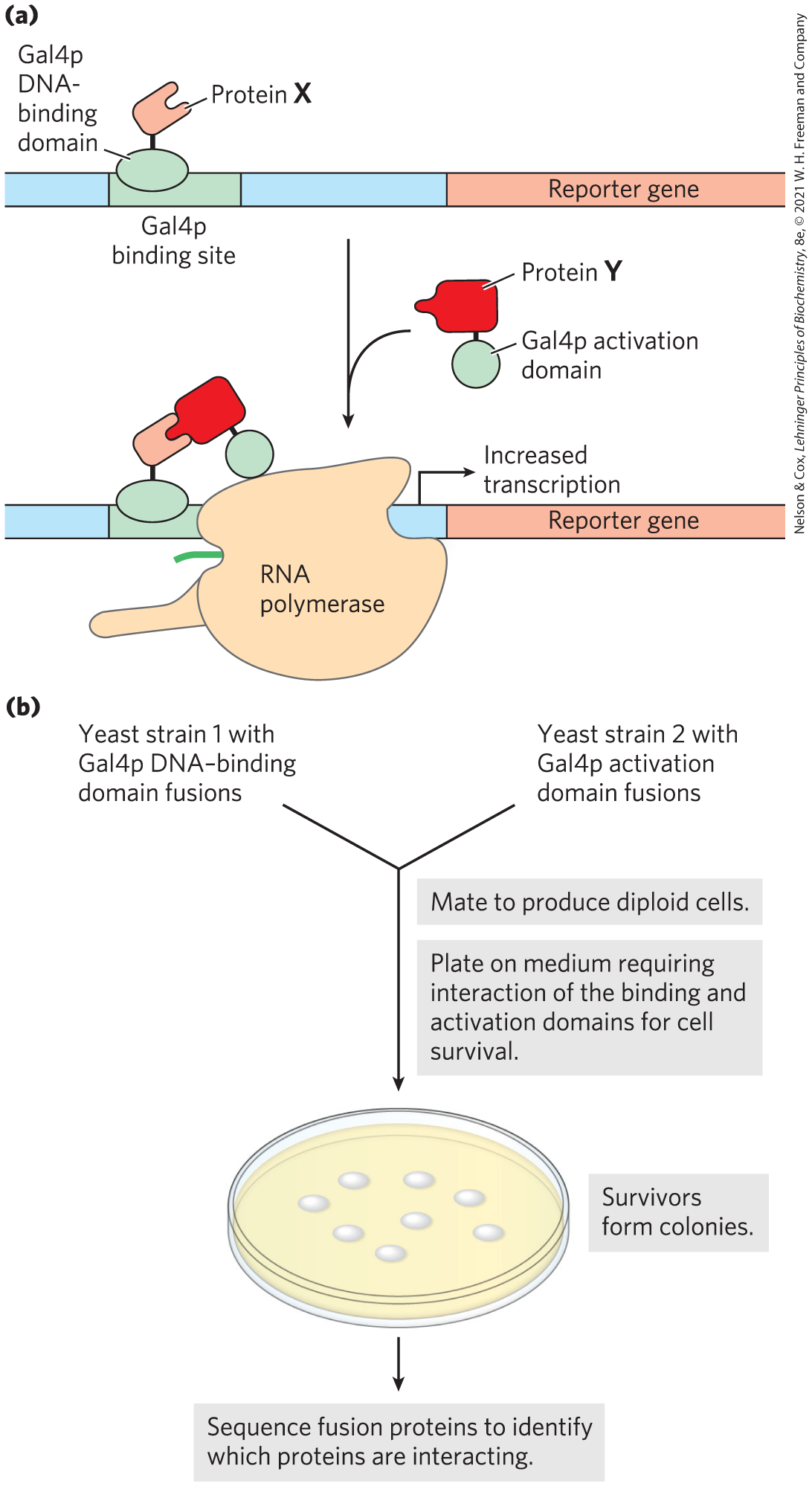
FIGURE 9-20 Yeast two-hybrid analysis. (a) The goal is to bring together the DNA-binding domain and the activation domain of the yeast Gal4 protein (Gal4p) through the interaction of two proteins, X and Y, to which one or other of the domains is fused. This interaction is accompanied by the expression of a reporter gene. (b) The two gene fusions are created in separate yeast strains, which are then mated. The mated mixture is plated on a medium on which the yeast cannot survive unless the reporter gene is expressed. Thus, all surviving colonies have interacting fusion proteins. Sequencing of the fusion proteins in the survivors reveals which proteins are interacting.
A library can be set up with a particular yeast strain in which each cell in the library has a gene fused to the Gal4p DNA-binding domain gene, and many such genes are represented in the library. In a second yeast strain, a gene of interest is fused to the gene for the Gal4p activation domain. The yeast strains are mated, and individual diploid cells are grown into colonies. The only cells that grow on the selective medium, or that produce the appropriate color, are those in which the gene of interest is binding to a partner, allowing transcription of the reporter gene. This allows large-scale screening for cellular proteins that interact with the target protein. The interacting protein that is fused to the Gal4p DNA-binding domain present in a particular selected colony can be quickly identified by DNA sequencing of the fusion protein’s gene. Some false positive results occur, due to the formation of multiprotein complexes.
The Effect of Deleting or Altering a Protein Can Suggest Its Function
One of the most informative paths to understanding the function of a gene is to change (mutate) the gene or delete it. An investigator can then examine how the genomic alteration affects cell growth or function. The methods available to modify genomes grow more sophisticated every year. The most common approach is to cut the gene of interest at a site that is functionally critical, generating a double-strand break. In eukaryotes, such breaks are most commonly repaired by cellular systems that promote nonhomologous end joining (NHEJ), a process described in Chapter 25. NHEJ seals the double-strand break, but the process is imprecise. Nucleotides are often deleted or added during the repair, inactivating the gene. In bacteria, introduced double-strand breaks are usually repaired more accurately, by homologous recombination systems (Chapter 25), but inactivating mutations can appear. Many traditional approaches to targeting a gene in this way were supplanted by the advent of CRISPR/Cas systems in 2011.
 One of the most informative paths to understanding the function of a gene is to change (mutate) the gene or delete it. An investigator can then examine how the genomic alteration affects cell growth or function. The methods available to modify genomes grow more sophisticated every year. The most common approach is to cut the gene of interest at a site that is functionally critical, generating a double-strand break. In eukaryotes, such breaks are most commonly repaired by cellular systems that promote nonhomologous end joining (NHEJ), a process described in
One of the most informative paths to understanding the function of a gene is to change (mutate) the gene or delete it. An investigator can then examine how the genomic alteration affects cell growth or function. The methods available to modify genomes grow more sophisticated every year. The most common approach is to cut the gene of interest at a site that is functionally critical, generating a double-strand break. In eukaryotes, such breaks are most commonly repaired by cellular systems that promote nonhomologous end joining (NHEJ), a process described in CRISPR/Cas Systems
“CRISPR” stands for clustered, regularly interspaced short palindromic repeats; as the name suggests, these consist of a series of regularly spaced short repeats in the bacterial genome. A Cas (CRISPR-associated) protein is a nuclease. The CRISPR sequences and Cas protein are components of a kind of immune system that evolved to allow bacteria to survive infection by bacteriophages. CRISPR sequences are embedded in the bacterial genome, surrounding sequences derived from phage pathogens that previously infected the bacterium without killing it. The viral sequences are, in effect, spacer sequences separating the CRISPR sequences. When the same bacteriophage again attacks a bacterium that has the corresponding CRISPR/Cas system, the CRISPR sequence and Cas protein act together to destroy the viral DNA. First, the CRISPR sequences are transcribed to RNA, and individual viral spacer sequences are cleaved to form products called guide RNAs (gRNAs), which include some adjacent repeat RNA. A gRNA forms a complex with one or more Cas proteins and, in some cases, with another RNA called a trans-activating CRISPR RNA, or tracrRNA. The resulting complex binds specifically to the invading bacteriophage DNA, cleaving and destroying it through the nuclease activities associated with the Cas proteins.
The current technology was made possible by discovery of a relatively simple CRISPR/Cas system in Streptococcus pyogenes. This system requires only a single Cas protein, Cas9, to cleave DNA. Work in many laboratories, particularly those of Jennifer Doudna and Emmanuelle Charpentier, has produced a streamlined CRISPR/Cas9 system composed of just one protein (Cas9) and one associated RNA, consisting of gRNA and tracrRNA fused into a single guide RNA (sgRNA). The power of the system is embedded in this sgRNA, in which the guide sequence can be altered to specifically and efficiently target almost any genomic sequence (Fig. 9-21). Cas9 has two separate nuclease domains: one domain cleaves the DNA strand paired with the sgRNA, and the other cleaves the opposite DNA strand. Inactivating one domain creates an enzyme that cleaves just one strand, forming a single-strand break, or nick. The sgRNA is needed both to pair with the target sequence in the DNA and to activate the nuclease domains for cleavage.
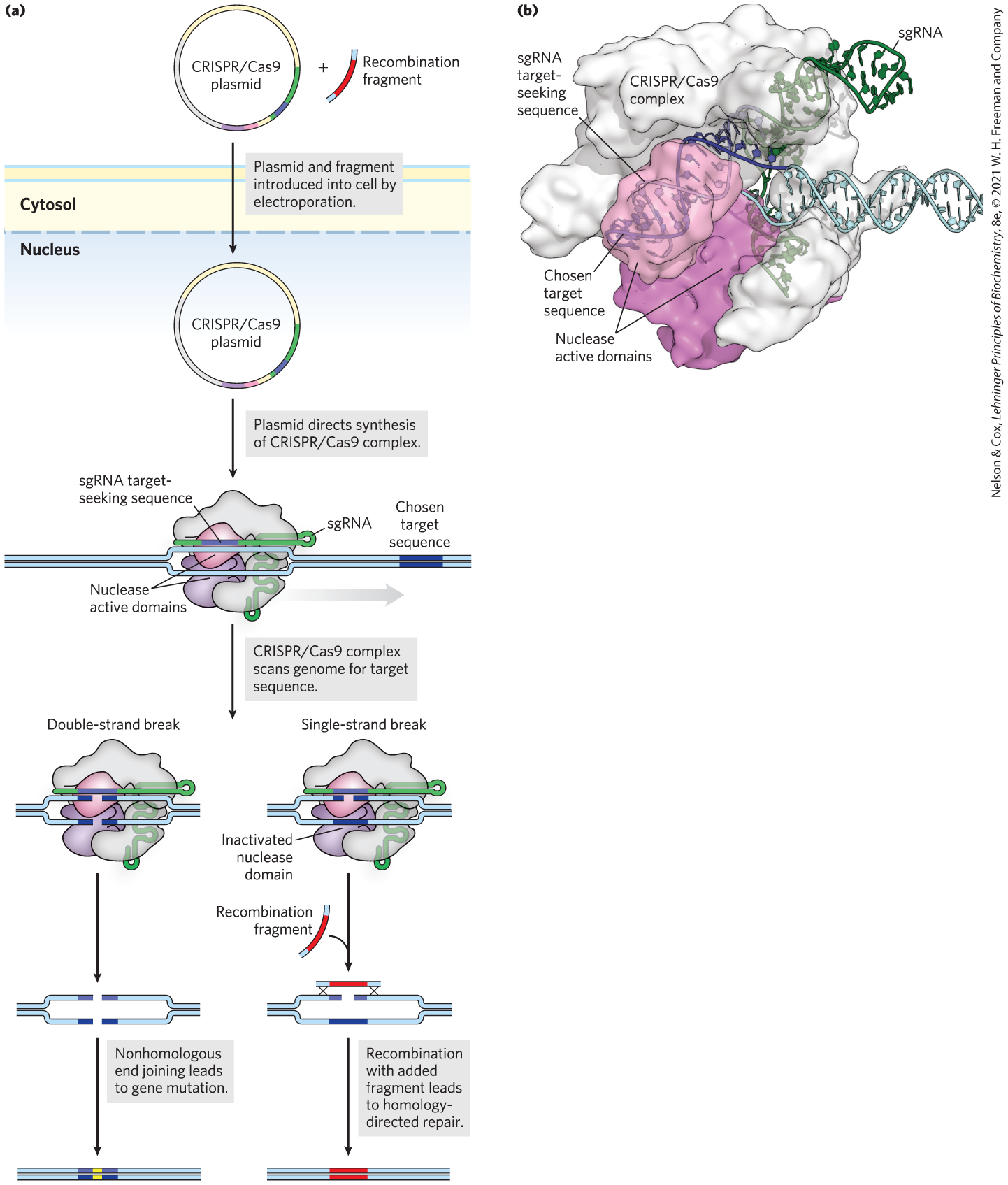
FIGURE 9-21 The CRISPR/Cas9 system for genomic engineering. (a) The genes encoding the Cas9 protein and sgRNA are introduced into a cell in which a targeted genomic change is planned. The sgRNA has a region complementary to the chosen genomic target sequence (purple); this region can be engineered to include any desired sequence. A complex consisting of the CRISPR sgRNA and the Cas9 protein forms within the cell and binds to the chosen target site in the DNA. The structure of the bound complex is shown in (b). In the pathway shown on the left in (a), two nuclease active sites in the Cas9 protein separately cleave each DNA strand in the target, producing a double-strand break. The double-strand break is usually repaired by nonhomologous end joining, which generally deletes or alters the nucleotides at the site where joining occurs. Alternatively, as shown in the pathway on the right, if one nuclease site is inactivated, Cas9 nuclease activity creates a single-strand break in the target sequence. In the presence of a recombination donor DNA fragment, identical to the target sequence but incorporating the desired sequence change (fragment shown in red), homologous DNA recombination will sometimes change the sequence at the site of the break to match that of the donor DNA. [Data from PDB ID 4UN3, C. Anders et al., Nature 513:569, 2014.]
Plasmids expressing the required protein and RNA components of CRISPR/Cas9 can be introduced into microbial cells by electroporation (p. 306). For mammalian cells, the genes encoding the CRISPR/Cas9 components can be incorporated into engineered viruses that subsequently deliver them to the cell nuclei. For many organisms, the targeted gene is inactivated in a high percentage of the treated cells. If a genomic change (mutation) rather than a simple gene inactivation is required, it can be introduced by recombination when a DNA fragment encompassing the cleavage site and including the desired change enters the cell with the CRISPR/Cas9 plasmids. This recombination is often inefficient, but success can be improved somewhat by introducing a nick rather than a double-strand break at the target site (Fig. 9-21).
CRISPR/Cas9 can be combined with other approaches to extract additional information. For example, a particular gene can be inactivated with CRISPR/Cas9. Then, the effect of that gene inactivation on the transcription of other genes can be probed with RNA-Seq at the level of tissues, cell populations, or single cells.
New applications for CRISPR/Cas9 are being developed rapidly, both for basic research and for medicine. Genetic screens based on CRISPR are described in the next section. CRISPR is being used to enhance food production, provide new approaches to combat bacterial infections, and eliminate nonnative pest species that can harbor diseases (Box 9-1). New CRISPR-based treatments for genetic diseases are being cautiously advanced to clinical trials for vision loss due to inherited retinal dystrophies, Duchenne muscular dystrophy, β-thalassaemia, and many other conditions. Uncertainties remain, particularly the potential for occasional cleavage at unintended chromosomal sites (off-target cleavage). The impact of CRISPR/Cas9 will continue to grow as problems are overcome, current applications mature, and new applications are imagined and created.
Many Proteins Are Still Undiscovered
For most biological processes, ranging from intermediary metabolism to neurological function to DNA metabolism, the list of known participating enzymes and proteins is far from complete. Genetic screening for new gene functions has been underway for many decades. The goal is to efficiently interrogate large numbers of genes, sometimes the entire genome, for genes that affect a particular cellular reaction or process. A gene perturbation — a treatment that inactivates a gene or activates its expression — is introduced under conditions in which just one gene is affected in each cell, but most or all of the genes are affected in one or more cells within the population (Fig. 9-22). The population is then subjected to a stress or selection. Cells in which a gene required to respond to the selection is altered may drop out of the population or be enriched in the population, depending upon the goals and design of the screen.
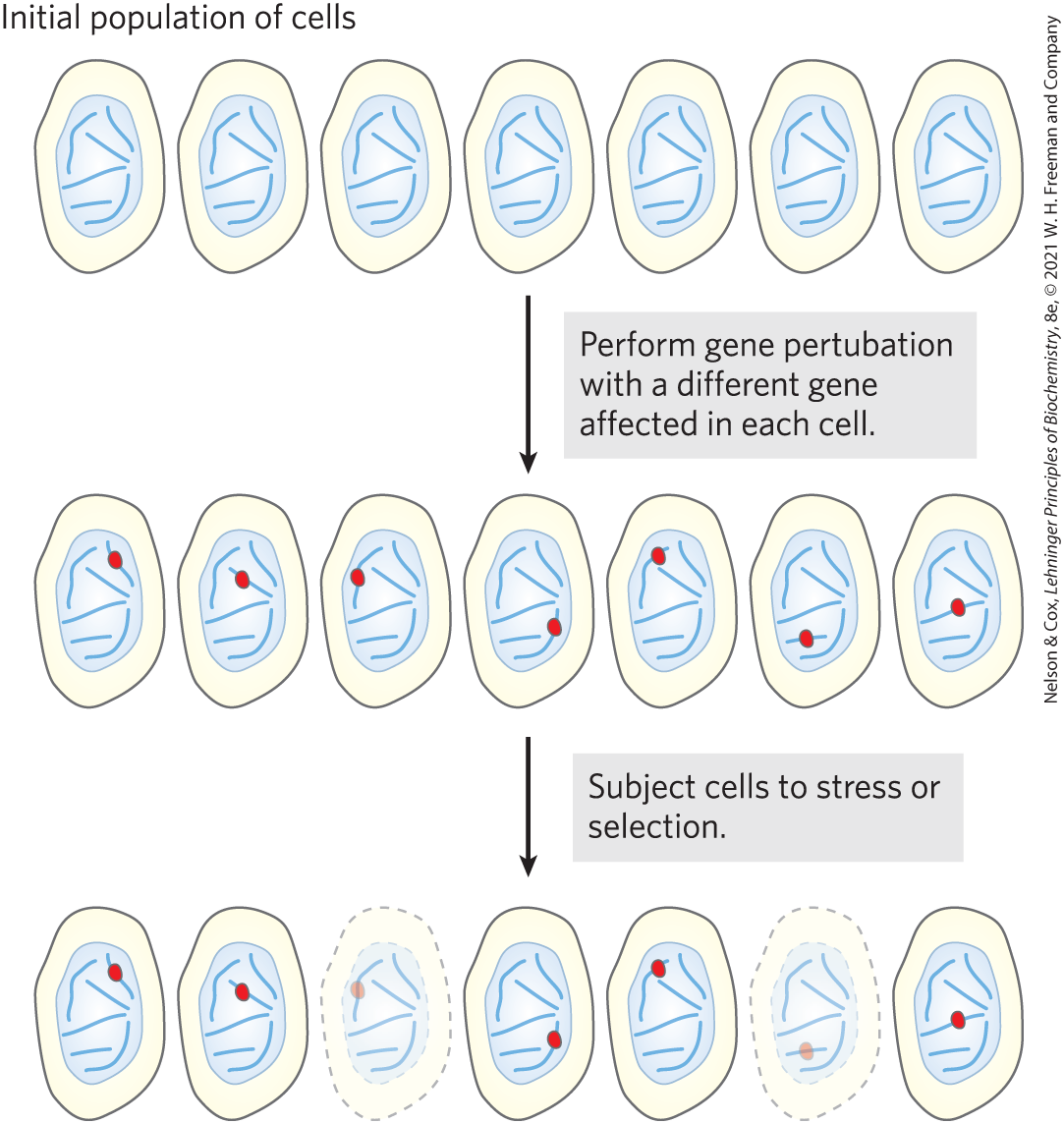
FIGURE 9-22 High-throughput genetic screening. A gene perturbation, either inactivation or activation, is introduced to a population of cells such that only one gene in each cell is affected. However, all or most genes are affected in one cell or another within the population. The population is then subjected to a selection that requires a response from some cellular genes. Cells that lack the required genes or that have those genes activated will drop out or be enriched within the population, respectively. In the example shown, two cells drop out.
CRISPR-based technologies increasingly play a central role in large-scale screening protocols (Fig. 9-23). Libraries of sgRNAs have been generated to target virtually all genes in a mammalian genome, or specialized subsets of them. The targeting sequence in each sgRNA is 20 bp long. In addition to targeting a particular gene, each targeting sequence acts as a kind of unique bar code identifier that is readily recognized by computer programs after sequencing. The sgRNAs are packaged in a DNA cassette set up to also express the Cas9 protein or a Cas9 variant. The cassettes are incorporated into carefully engineered lentiviral vectors derived from HIV (with genes required for HIV multiplication eliminated). The viral vectors deliver the cassette to the nucleus as a single-stranded RNA, convert it to double-stranded DNA with the viral-encoded reverse transcriptase, and integrate the DNA into a chromosome. The CRISPR/Cas9 components are expressed to perturb the target gene specified by the particular sgRNA delivered to that cell. The effect produced depends upon the Cas9 variant used. The unmodified Cas9 nuclease will create a double-strand break that inactivates the gene. A modified Cas9 that lacks the nuclease activity will simply bind to its target and block transcription. Cas9 fused to a protein transcription inhibitor or activator may more effectively block or activate transcription, respectively (Fig. 9-23b).
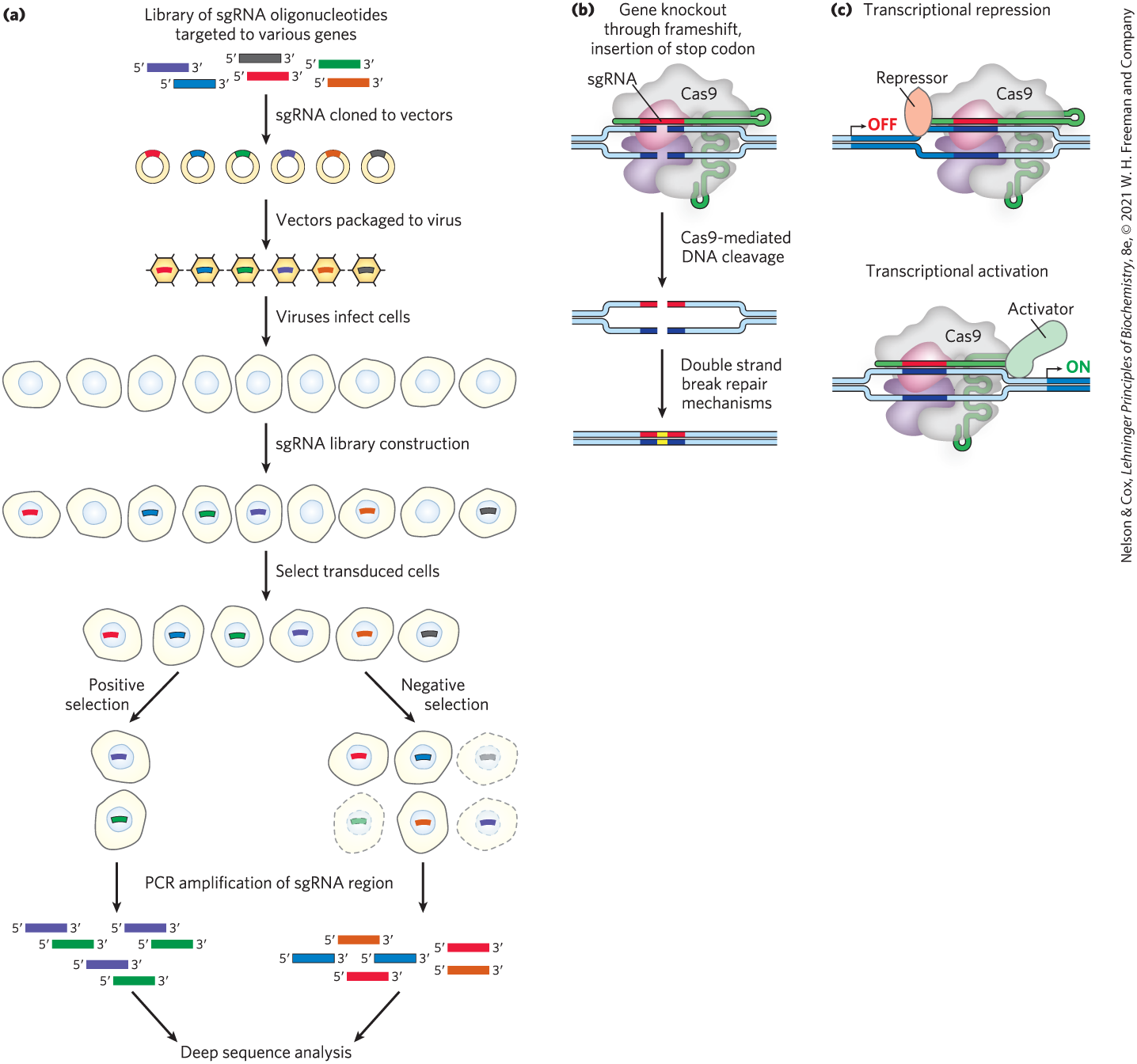
FIGURE 9-23 Use of CRISPR/Cas9 in high-throughput screening. CRISPR/Cas9 provides the gene perturbation in many screening protocols. (a) In a typical screen, a library of sgRNAs is constructed so as to target all known genes in a genome of interest. These are cloned into viral vectors. The vectors infect cells at a multiplicity of infection (MOI) small enough so that most cells will gain only one vector. The vector RNA is converted to DNA and integrated into the genome. When expressed, it will affect one target gene, with most genes affected in one or more cells in the population. After selection, some cells drop out or are enriched in the population, depending on the nature of the screen. (b) Several variations of Cas9 are shown to illustrate a few of the ways genes can be affected. Unaltered Cas9 will cleave the DNA at the target site. (c) If engineered to lack nuclease activity, and fused with a gene repressor or activator, the modified Cas9 will bind to the target site and either decrease or increase gene transcription, respectively.
Whatever strategy is used, a different gene is affected in each cell. Once the population has been treated with a stress or selection, cells in which genes required to survive the treatment are inactivated or activated by the CRISPR/Cas9 variant will die or thrive. The decreased or increased presence of the relevant bar code sequences can be detected by deep DNA sequencing, using a universal priming sequence incorporated into the cassette near the sgRNA sequence. The strategies outlined here only hint at the variety of protocols in use, limited only by the imagination of the investigators.
SUMMARY 9.2 Exploring Protein Function on the Scale of Cells or Whole Organisms
- Proteins can be studied at the level of phenotypic, cellular, or molecular function.
- Comparative genomics can elucidate protein function by identifying structural motifs within the encoded protein and comparing gene sequences from different organisms.
- A determination of when and where a protein appears in a cell can offer functional clues. RNA-Seq provides information on what genes are being expressed in a cell. Mass spectrometry can define cellular proteomes. By fusing a gene of interest with genes that encode green fluorescent protein or epitope tags, researchers can visualize the cellular location of the gene product, either directly or by immunofluorescence.
- The interactions of a protein with other proteins or RNA can be investigated with epitope tags and immunoprecipitation or affinity chromatography. Yeast two-hybrid analysis probes molecular interactions in vivo.
- The cellular effects of inactivating a gene can be conveniently explored using the CRISPR/Cas9 programmable nuclease. CRISPR/Cas9 can also be used to alter gene sequences in a targeted manner.
- Screens for new genes increasingly employ variants of the CRISPR/Cas9 system.
 Proteins can be studied at the level of phenotypic, cellular, or molecular function.
Proteins can be studied at the level of phenotypic, cellular, or molecular function.