3.3 Working with Proteins
Biochemists’ understanding of protein structure and function has been derived from the study of many individual proteins. To study a protein in detail, the researcher must be able to separate it from other proteins in pure form and must have the techniques to determine its properties. The necessary methods come from protein chemistry, a discipline as old as biochemistry itself and one that retains a central position in biochemical research.
Proteins Can Be Separated and Purified
A pure preparation is usually essential before a protein’s properties and activities can be determined. Given that cells contain thousands of different kinds of proteins, how can one protein be purified? Methods for separating proteins take advantage of properties that vary from one protein to the next, including size, charge, and binding properties. The advent of genetic engineering approaches has provided new and simpler paths for protein purification. The latter methods, described in Chapter 9, often artificially modify the protein being purified, adding a few or many amino acid residues to one or both ends. In many cases, the modifications alter protein function. Isolation of unaltered native proteins requires removal of the modification or a reliance on methods described here.
 Methods for separating proteins take advantage of properties that vary from one protein to the next, including size, charge, and binding properties. The advent of genetic engineering approaches has provided new and simpler paths for protein purification. The latter methods, described in
Methods for separating proteins take advantage of properties that vary from one protein to the next, including size, charge, and binding properties. The advent of genetic engineering approaches has provided new and simpler paths for protein purification. The latter methods, described in The source of a protein is generally tissue or microbial cells. The first step in any protein purification procedure is to break open these cells, releasing their proteins into a solution called a crude extract. If necessary, differential centrifugation can be used to prepare subcellular fractions or to isolate specific organelles (see Fig. 1-7).
Once the extract or organelle preparation is ready, various methods are available for purifying one or more of the proteins it contains. Commonly, the extract is subjected to treatments that separate the proteins into different fractions based on a property such as size or charge; the process is referred to as fractionation. Early fractionation steps in a purification utilize differences in protein solubility, which is a complex function of pH, temperature, salt concentration, and other factors. The solubility of proteins is lowered in the presence of some salts, an effect called “salting out.” Ammonium sulfate is particularly effective for selectively precipitating some proteins while leaving others in solution. Low-speed centrifugation is then used to remove the precipitated proteins from those remaining in solution.
A solution containing the protein of interest usually must be further altered before subsequent purification steps are possible. For example, dialysis is a procedure that separates proteins from small solutes by taking advantage of the proteins’ larger size. The partially purified extract is placed in a bag or tube made of a semipermeable membrane, which is suspended in a much larger volume of buffered solution of appropriate ionic strength. The membrane allows the exchange of salt and buffer but not proteins. Thus dialysis retains large proteins within the membranous bag or tube while allowing the concentration of other solutes in the protein preparation to change until they come into equilibrium with the solution outside the membrane. Dialysis might be used, for example, to remove ammonium sulfate from the protein preparation.
The most efficient methods for fractionating proteins make use of column chromatography, which takes advantage of differences in protein charge, size, binding affinity, and other properties (Fig. 3-16). A porous solid material with appropriate chemical properties (the stationary phase) is held in a column, and a buffered solution (the mobile phase) migrates through it. The protein, dissolved in the same buffered solution that was used to establish the mobile phase, is layered on the top of the column. The protein then percolates through the solid matrix as an ever-expanding band within the larger mobile phase. Individual proteins migrate faster or more slowly through the column, depending on their properties.
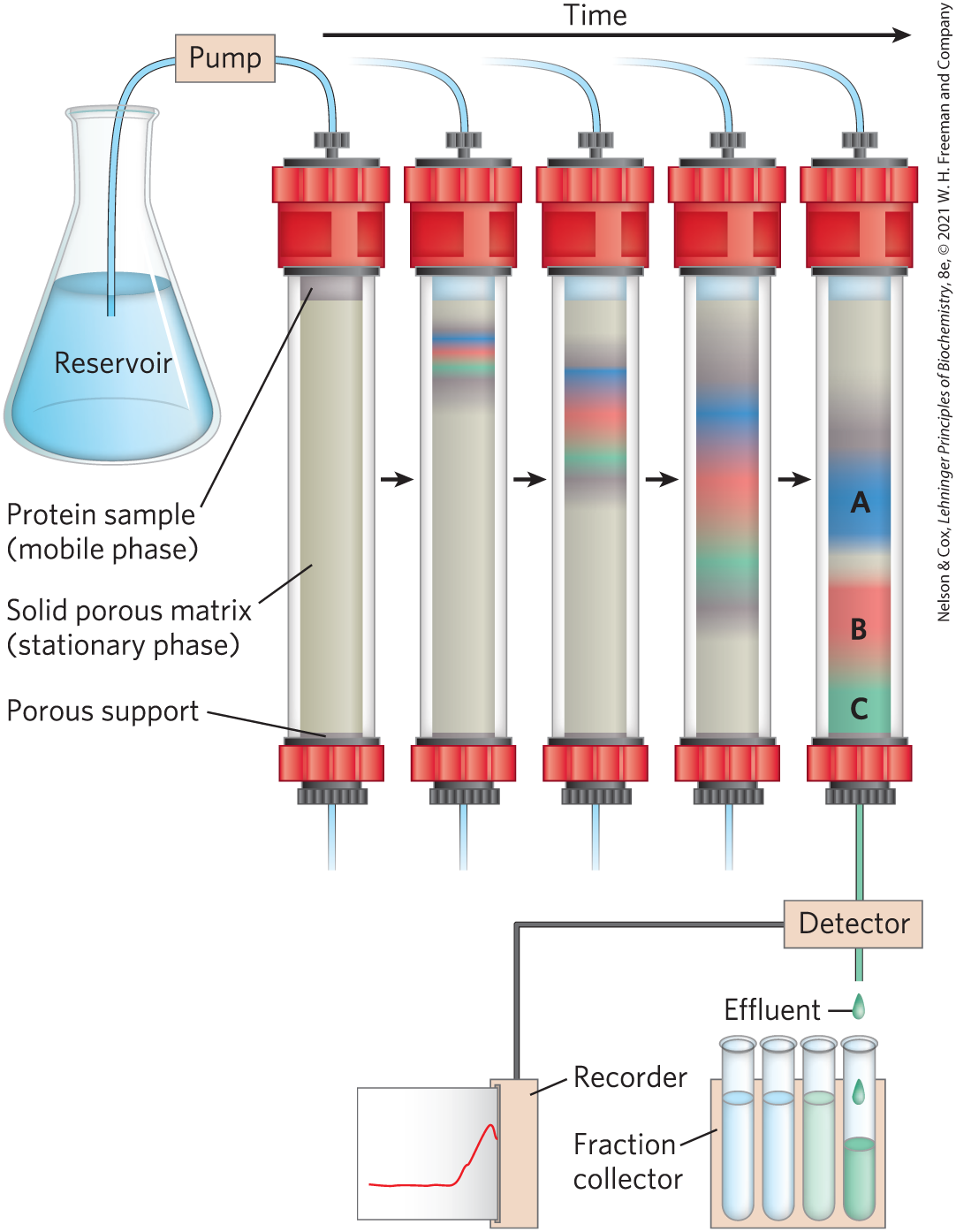
FIGURE 3-16 Column chromatography. The standard elements of a chromatographic column include a solid, porous material (matrix) supported inside a column, generally made of plastic or glass. A solution, the mobile phase, flows through the matrix, the stationary phase. The solution that passes out of the column at the bottom (the effluent) is constantly replaced by solution supplied from a reservoir at the top. The protein solution to be separated is layered on top of the column and allowed to percolate into the solid matrix. Additional solution is added on top. The protein solution forms a band within the mobile phase that is initially the depth of the protein solution applied to the column. As proteins migrate through the column (shown here at five different times), they are retarded to different degrees by their different interactions with the matrix material. The overall protein band thus widens as it moves through the column. Individual types of proteins (such as A, B, and C, shown in blue, red, and green) gradually separate from each other, forming bands within the broader protein band. Separation improves (i.e., resolution increases) as the length of the column increases. However, each individual protein band also broadens with time due to diffusional spreading, a process that decreases resolution. In this example, protein A is well separated from B and C, but diffusional spreading prevents complete separation of B and C under these conditions. Protein C is being detected and its presence recorded as it is eluted from the column.
Ion-exchange chromatography exploits differences in the sign and magnitude of the net electric charge of proteins at a given pH (Fig. 3-17a). The column matrix is a synthetic polymer (resin) containing bound charged groups; those with bound anionic groups are called cation exchangers, and those with bound cationic groups are called anion exchangers. The affinity of each protein for the charged groups on the column is affected by the pH (which determines the ionization state of the molecule) and the concentration of competing free salt ions in the surrounding solution. Separation can be optimized by gradually changing the pH and/or salt concentration of the mobile phase in order to create a pH or salt gradient. In cation-exchange chromatography, proteins with a net positive charge migrate through the matrix more slowly than those with a net negative charge, because the migration of the former is retarded more by interaction with the stationary phase.
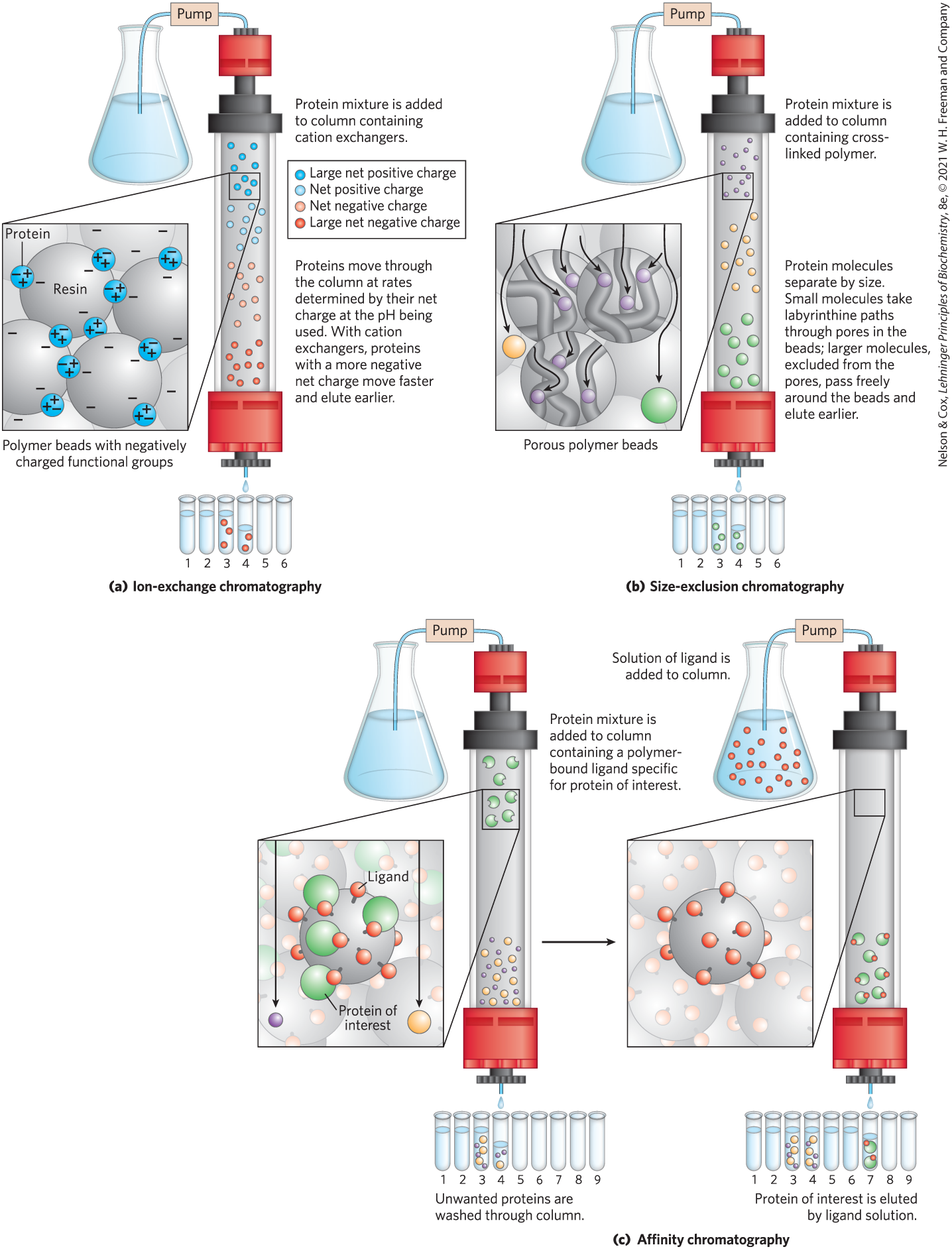
FIGURE 3-17 Three chromatographic methods used in protein purification. (a) Ion-exchange chromatography exploits differences in the sign and magnitude of the net electric charges of proteins at a given pH. (b) Size-exclusion chromatography, also called gel filtration, separates proteins according to size. (c) Affinity chromatography separates proteins by their binding specificities. Further details of these methods are given in the text.
As the protein-containing solution exits a column, successive portions (fractions) of this effluent are collected in test tubes. Each fraction can be tested for the presence of the protein of interest as well as other properties, such as ionic strength or total protein concentration. All fractions positive for the protein of interest can be combined as the product of this chromatographic step of the protein purification.
WORKED EXAMPLE 3-1 Ion Exchange of Peptides
A biochemist wants to separate two peptides by ion-exchange chromatography. At the pH of the mobile phase to be used on the column, one peptide (A) has a pI of 5.1, due to the presence of more Glu and Asp residues than Arg, Lys, and His residues, and has a net negative charge at neutral pH. Peptide B has a pI of 7.8, reflecting a plurality of positively charged amino acid residues at neutral pH. At neutral pH, which peptide would elute first from a cation-exchange resin? Which would elute first from an anion-exchange resin?
SOLUTION:
A cation-exchange resin has negative charges and binds positively charged molecules, retarding their progress through the column. Peptide B, with its higher pI and net positive charge, will interact more strongly than peptide A with the cation-exchange resin. Thus, peptide A will elute first. On the anion-exchange resin, peptide B will elute first. Peptide A, having a relatively low pI and a net negative charge, will be retarded by its interaction with the positively charged resin.
Figure 3-17 shows two variations of column chromatography in addition to ion exchange. Size-exclusion chromatography, also called gel filtration (Fig. 3-17b), separates proteins according to size. In this method, large proteins emerge from the column sooner than small ones — a somewhat counterintuitive result. The solid phase consists of cross-linked polymer beads with engineered pores or cavities of a particular size. Large proteins cannot enter the cavities and so take a shorter (and more rapid) path through the column, around the beads. Small proteins enter the cavities and are slowed by their more labyrinthine path through the column. Size-exclusion chromatography can also be used to approximate the size of a protein being purified, using methods similar to those described in Figure 3-19.
Affinity chromatography is based on binding affinity (Fig. 3-17c). The beads in the column have a covalently attached chemical group called a ligand — a group or molecule that binds to a macromolecule such as a protein. When a protein mixture is added to the column, any protein with affinity for this ligand binds to the beads, and its migration through the matrix is retarded. For example, if the biological function of a protein involves binding to ATP, then attaching a molecule that resembles ATP to the beads in the column creates an affinity matrix that can help purify the protein. Proteins that do not bind to ATP flow more rapidly through the column. Bound proteins are then eluted by a solution containing either a high concentration of salt or a free ligand — in this case, ATP or an analog of ATP. Salt weakens the binding of the protein to the immobilized ligand, interfering with ionic interactions. Free ligand competes with the ligand attached to the beads, releasing the protein from the matrix; the protein product that elutes from the column is often bound to the ligand used to elute it.
Protein purification protocols often use genetic engineering to fuse additional amino acids or peptides (tags) to the target protein. Affinity chromatography can be used to bind this tag, achieving a large increase in purity in a single step (see Fig. 9-11). In many cases, the tag can be subsequently removed, fully restoring the function of the native protein.
Chromatographic methods are typically enhanced by the use of HPLC, or high-performance liquid chromatography. HPLC makes use of high-pressure pumps that speed the movement of the protein molecules down the column; it also uses higher-quality chromatographic materials that can withstand the crushing force of the pressurized flow. By reducing the transit time on the column, HPLC can limit diffusional spreading of protein bands and thus can greatly improve resolution.
Choosing the approach to purification of a protein that has not previously been isolated is guided both by established precedents and by common sense. In most cases, several different methods must be used sequentially to purify a protein completely, each separating proteins on the basis of different properties. The choice of methods is somewhat empirical, and many strategies may be tried before the most effective one is found. Researchers can often minimize trial and error by basing the new procedure on purification techniques developed for similar proteins. Common sense dictates that inexpensive procedures such as salting out be used first, when the total volume and the number of contaminants are greatest. As each purification step is completed, the sample size generally becomes smaller (Table 3-5), making it feasible to use more sophisticated (and expensive) chromatographic procedures at later stages. A purification table documents the success of each step in a purification protocol. In the hypothetical purification shown in Table 3-5, the ratio of the final specific activity (15,000 units/mg) to the starting specific activity (10 units/mg) gives the purification factor (1,500). The percentage of the total activity at the last step (45,000 units) relative to the total activity in the starting material (100,000 units) gives the yield from the purification procedure (45%).
| Procedure or step | Fraction volume (mL) | Total protein (mg) | Activity (units) | Specific activity (units/mg) |
|---|---|---|---|---|
1. Crude cellular extract |
1,400 |
10,000 |
100,000 |
10 |
2. Precipitation with ammonium sulfate |
280 |
3,000 |
96,000 |
32 |
3. Ion-exchange chromatography |
90 |
400 |
80,000 |
200 |
4. Size-exclusion chromatography |
80 |
100 |
60,000 |
600 |
5. Affinity chromatography |
6 |
3 |
45,000 |
15,000 |
|
Note: All data represent the status of the sample after the designated procedure has been carried out. “Activity” and “specific activity” are defined on page 90. |
||||
Proteins Can Be Separated and Characterized by Electrophoresis
Protein purification is usually complemented by electrophoresis, an analytical process that allows researchers to visualize and characterize proteins as they are purified. This method does not itself contribute to purification, as electrophoresis often adversely affects the structure and thus the function of proteins. However, it allows a biochemist to rapidly estimate the number of different proteins in a mixture and the degree of purity of a particular protein preparation. Also, electrophoresis can be used to determine such crucial properties of a protein as its isoelectric point and approximate molecular weight.
Electrophoresis of proteins is generally carried out in gels made up of the cross-linked polymer polyacrylamide (Fig. 3-18). The polyacrylamide gel acts as a molecular sieve, slowing the migration of proteins approximately in proportion to their charge-to-mass ratio. Migration may also be affected by protein shape. In electrophoresis, the force moving the macromolecule is the electrical potential, E. The electrophoretic mobility, μ, of a molecule is the ratio of its velocity, V, to the electrical potential. Electrophoretic mobility is also equal to the net charge, Z, of the molecule divided by the frictional coefficient, f, which reflects in part a protein’s shape. Thus,
The migration of a protein in a gel during electrophoresis is therefore a function of its size and its shape.
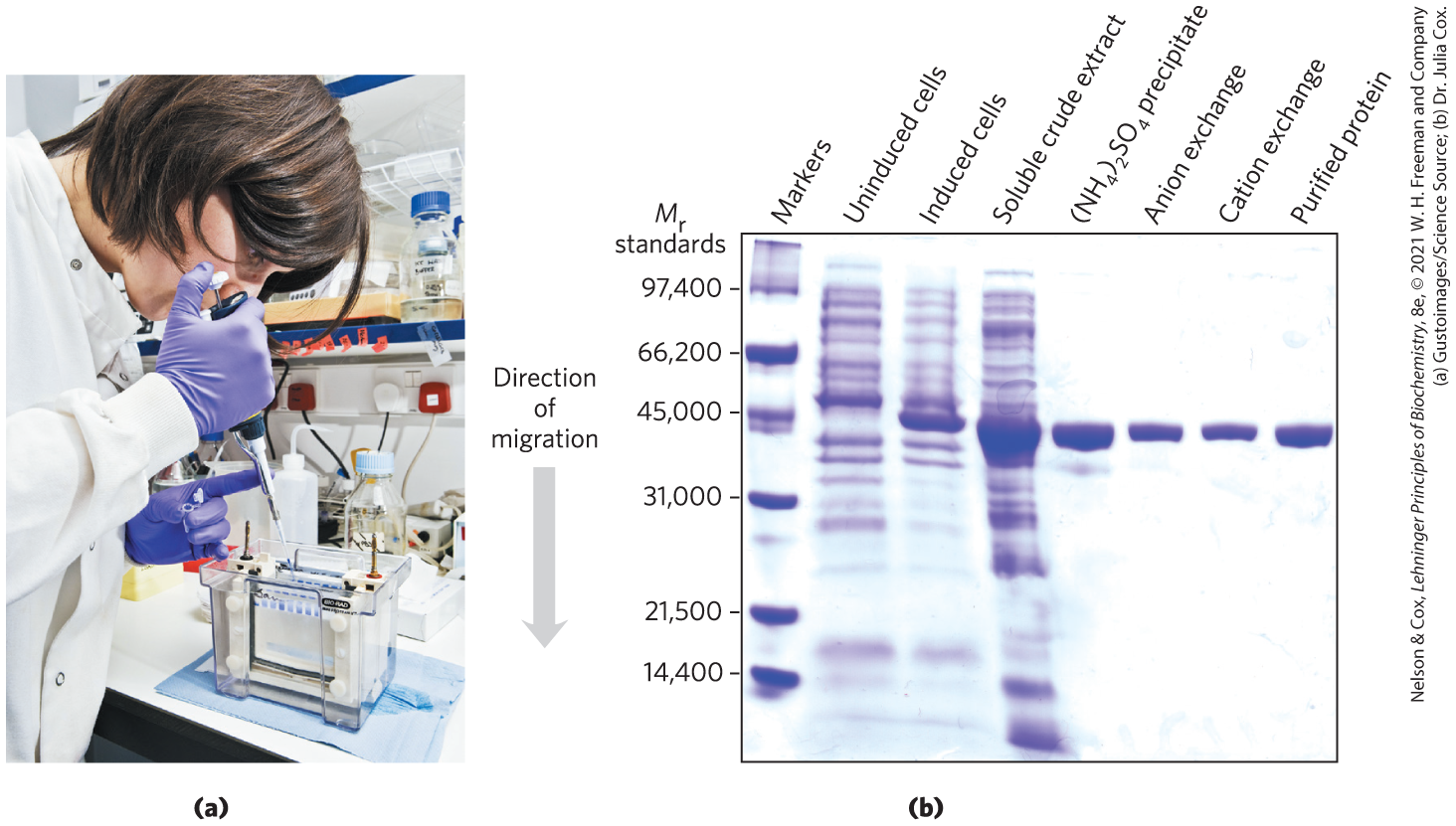
FIGURE 3-18 Electrophoresis. (a) Different samples are loaded in wells or depressions at the top of the SDS polyacrylamide gel. The proteins move into the gel when an electric field is applied. The gel minimizes convection currents caused by small temperature gradients, as well as protein movements other than those induced by the electric field. (b) Proteins can be visualized after electrophoresis by treating the gel with a stain such as Coomassie blue, which binds to the proteins but not to the gel itself. Each band on the gel represents a different protein (or protein subunit); smaller proteins move through the gel more rapidly than larger proteins and therefore are found nearer the bottom of the gel. This gel illustrates purification of the RecA protein of Escherichia coli. The gene for the RecA protein was cloned so that its expression (synthesis of the protein) could be controlled. The first lane shows a set of standard proteins (of known ), serving as molecular weight markers. The second and third lanes show, respectively, proteins from E. coli cells before and after synthesis of RecA protein was induced. The fourth lane shows the proteins in a crude cellular extract. Subsequent lanes (left to right) show the proteins that are present after successive purification steps. Although the protein looks pure in lane 6, two more steps are needed to remove minor contaminants not evident on the gel. The purified protein is a single polypeptide chain as seen in the rightmost lane.
The electrophoretic method commonly employed for estimation of purity and molecular weight makes use of the detergent sodium dodecyl sulfate (SDS) (“dodecyl” denoting a 12-carbon chain).
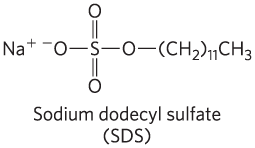
A protein will bind about 1.4 times its weight of SDS, nearly one molecule of SDS for each amino acid residue. The sulfate moieties of the bound SDS contribute a large net negative charge, rendering the intrinsic charge of the protein insignificant and conferring on each protein a similar charge-to-mass ratio. In addition, SDS binding partially unfolds proteins, such that most SDS-bound proteins assume a similar rodlike shape. Electrophoresis in the presence of SDS therefore separates proteins almost exclusively on the basis of mass (molecular weight), with smaller polypeptides migrating more rapidly. After electrophoresis, the proteins are visualized by adding a dye such as Coomassie blue, which binds to proteins but not to the gel itself (Fig. 3-18b). Thus, a researcher can monitor the progress of a protein purification procedure as the number of protein bands visible on the gel decreases after each new fractionation step. When compared with the positions to which proteins of known molecular weight migrate in the gel, the position of an unidentified protein can provide a good approximation of its molecular weight (Fig. 3-19). If the protein has two or more different subunits, generally the subunits are separated by the SDS treatment, and a separate band appears for each.
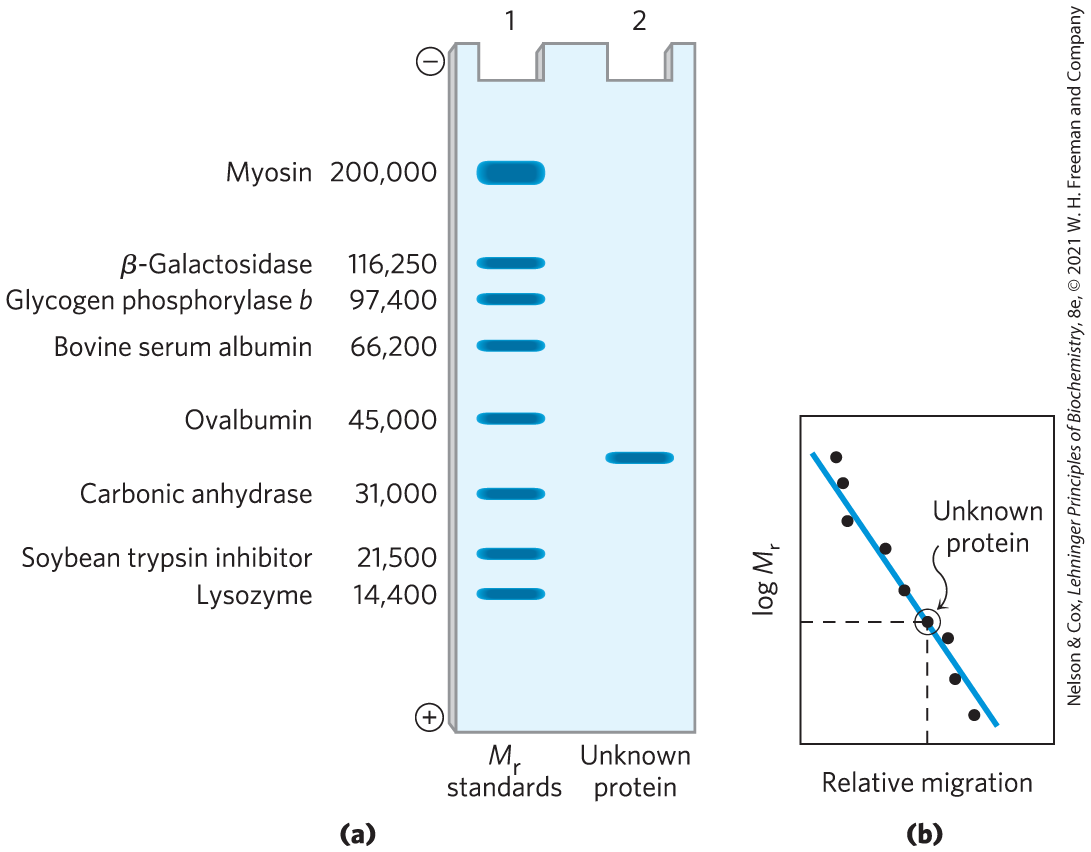
FIGURE 3-19 Estimating the molecular weight of a protein. The electrophoretic mobility of a protein on an SDS polyacrylamide gel is related to its molecular weight, . (a) Standard proteins of known molecular weight are subjected to electrophoresis (lane 1). These marker proteins can be used to estimate the molecular weight of an unknown protein (lane 2). (b) A plot of of the marker proteins versus relative migration during electrophoresis is linear, which allows the molecular weight of the unknown protein to be read from the graph. (In similar fashion, a set of standard proteins with reproducible retention times on a size-exclusion column can be used to create a standard curve of retention time versus log . The retention time of an unknown substance on the column can be compared with this standard curve to obtain an approximate .)
Isoelectric focusing is a procedure used to determine the isoelectric point (pI) of a protein (Fig. 3-20). A pH gradient is established by allowing a mixture of low molecular weight organic acids and bases (ampholytes; p. 77) to distribute themselves in an electric field generated across the gel. When a protein mixture is applied, each protein migrates until it reaches the pH that matches its pI. Proteins with different isoelectric points are thus distributed differently throughout the gel.
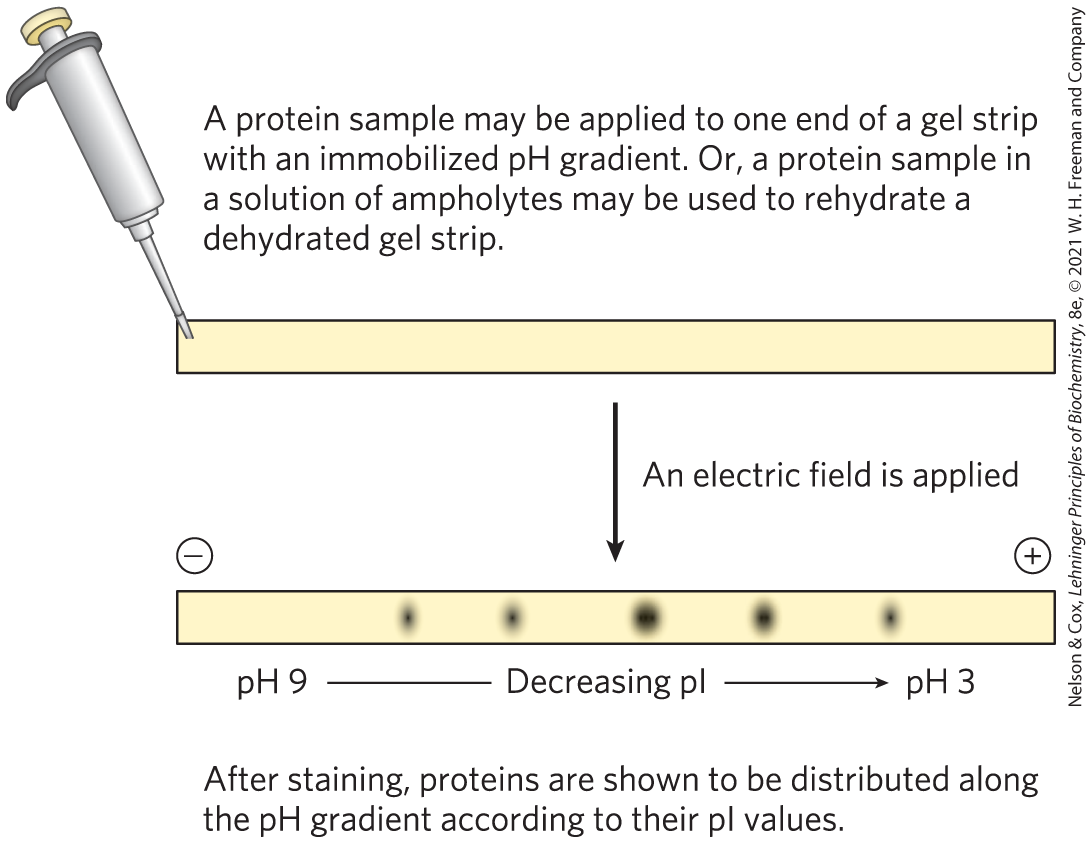
FIGURE 3-20 Isoelectric focusing. This technique separates proteins according to their isoelectric points. A protein mixture is placed on a gel strip containing an immobilized pH gradient. With an applied electric field, proteins enter the gel and migrate until each reaches a pH that is equivalent to its pI. Remember that when pH = pI, the net charge of a protein is zero.
Combining isoelectric focusing and SDS electrophoresis sequentially in a process called two-dimensional electrophoresis permits the resolution of complex mixtures of proteins (Fig. 3-21). This is a more sensitive analytical method than either electrophoretic method alone. Two-dimensional electrophoresis separates proteins of identical molecular weight that differ in pI, or proteins with similar pI values but different molecular weights.
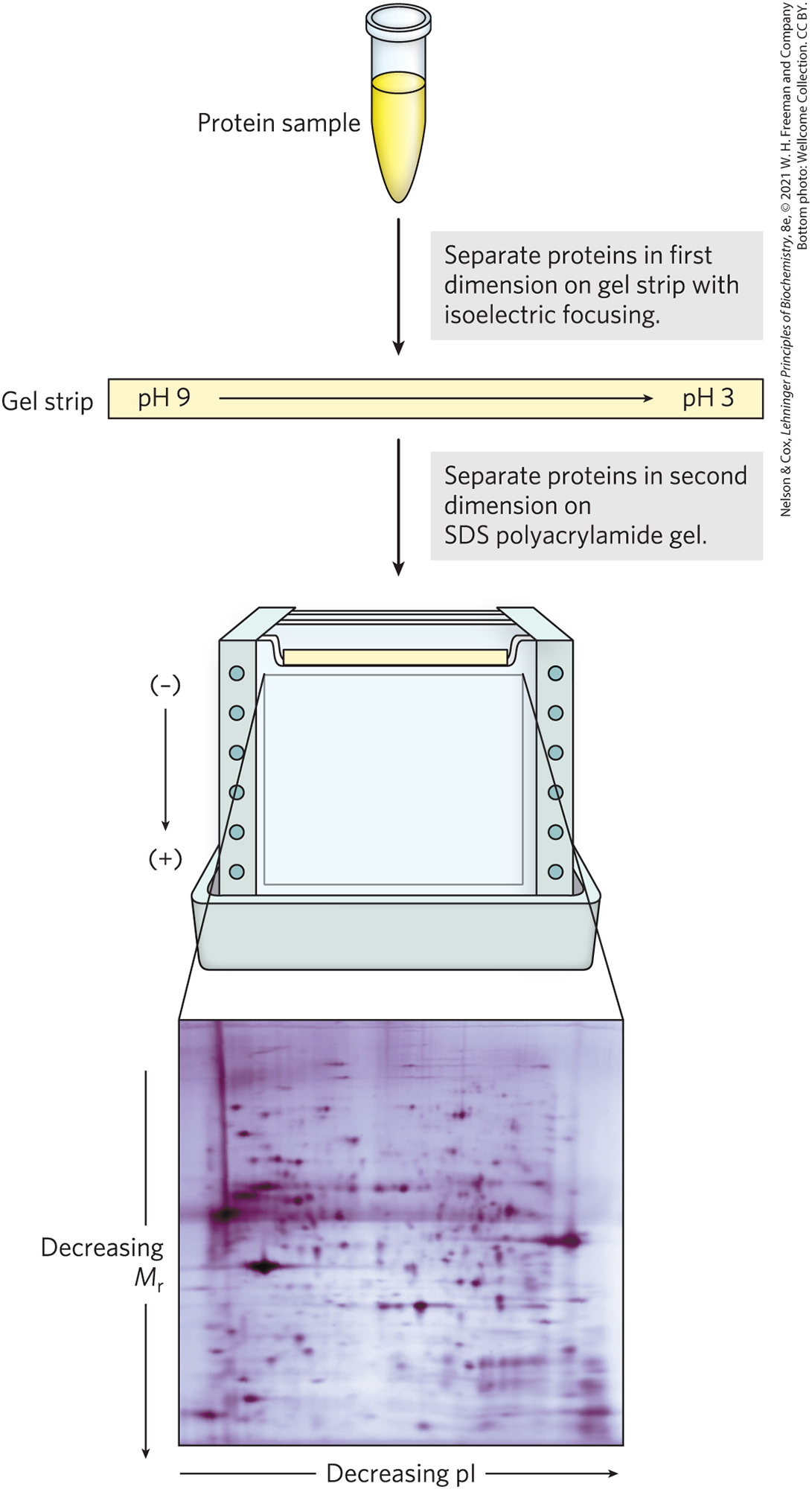
FIGURE 3-21 Two-dimensional electrophoresis. Proteins are first separated by isoelectric focusing in a thin strip gel. The gel is then laid horizontally on a second, slab-shaped gel, and the proteins are separated by SDS polyacrylamide gel electrophoresis. Horizontal separation reflects differences in pI; vertical separation reflects differences in molecular weight. The original protein complement is thus spread in two dimensions. Thousands of cellular proteins can be resolved using this technique. Individual protein spots can be cut out of the gel and identified by mass spectrometry (see Figs 3-28 and 3-29).
Unseparated Proteins Are Detected and Quantified Based on Their Functions
To purify a protein, it is essential to have a way of detecting and quantifying that protein in the presence of many other proteins at each stage of the procedure. A common target of purification is one or another of the class of proteins called enzymes (Chapter 6). Each enzyme catalyzes a particular reaction that converts one biomolecule (the substrate) to another (the product). The amount of the protein in a given solution or tissue extract can be measured, or assayed, in terms of the catalytic effect the enzyme produces — that is, the increase in the rate at which its substrate is converted to reaction products when the enzyme is present. For this purpose the researcher must know (1) the overall equation of the reaction catalyzed, (2) an analytical procedure for determining the disappearance of the substrate or the appearance of a reaction product, (3) whether the enzyme requires cofactors such as metal ions or coenzymes, (4) the dependence of the enzyme activity on substrate concentration, (5) the optimum pH, and (6) a temperature zone in which the enzyme is stable and has high activity. Enzymes are usually assayed at their optimum pH and at some convenient temperature within the range 25 to . Also, very high substrate concentrations are generally used so that the initial reaction rate, measured experimentally, is proportional to enzyme concentration (Chapter 6).
By international agreement, 1.0 unit of enzyme activity for most enzymes is defined as the amount of enzyme causing transformation of 1.0 μmol of substrate to product per minute at under optimal conditions of measurement (for many enzymes, this definition is inconvenient, and a unit may be defined differently). The term activity refers to the total units of enzyme in a solution. The specific activity is the number of enzyme units per milligram of total protein (Fig. 3-22). The specific activity is a measure of enzyme purity: it increases during purification of an enzyme and becomes maximal and constant when the enzyme is pure (Table 3-5).

FIGURE 3-22 Activity versus specific activity. The difference between these terms can be illustrated by considering two flasks containing marbles. The flasks contain the same number of red marbles, but different numbers of marbles of other colors. If the marbles represent proteins, both flasks contain the same activity of the protein, represented by the red marbles. The second flask, however, has the higher specific activity, because red marbles represent a higher fraction of the total.
After each purification step, the activity of the preparation (in units of enzyme activity) is assayed, the total amount of protein is determined independently, and the ratio of the two gives the specific activity. Activity and total protein generally decrease with each step. Activity decreases because there is always some loss due to inactivation or nonideal interactions with chromatographic materials or other molecules in the solution. Total protein decreases because the objective is to remove as much unwanted or nonspecific protein as possible. In a successful step, the loss of nonspecific protein is much greater than the loss of activity; therefore, specific activity increases even as total activity falls. The data are assembled in a purification table similar to Table 3-5. A protein is generally considered pure when further purification steps fail to increase specific activity and when only a single protein species can be detected (for example, by electrophoresis in the presence of SDS).
For proteins that are not enzymes, other quantification methods are required. Transport proteins can be assayed by their binding to the molecule they transport, and hormones and toxins by the biological effect they produce; for example, growth hormones will stimulate the growth of certain cultured cells. Some structural proteins represent such a large fraction of a tissue mass that they can be readily extracted and purified without a functional assay. The approaches are as varied as the proteins themselves.
SUMMARY 3.3 Working with Proteins
- Proteins are separated and purified on the basis of differences in their properties. Proteins can be selectively precipitated by changes in pH or temperature, and particularly by the addition of certain salts. A wide range of chromatographic procedures makes use of differences in size, binding affinities, charge, and other properties. These include ion-exchange, size-exclusion, affinity, and high-performance liquid chromatography.
- Electrophoresis separates proteins on the basis of mass or charge for analytical purposes. SDS gel electrophoresis and isoelectric focusing can be used separately or in combination for higher resolution.
- All purification procedures require a method for quantifying or assaying the protein of interest in the presence of other proteins. Purification can be monitored by assaying specific activity.
 Proteins are separated and purified on the basis of differences in their properties. Proteins can be selectively precipitated by changes in pH or temperature, and particularly by the addition of certain salts. A wide range of chromatographic procedures makes use of differences in size, binding affinities, charge, and other properties. These include ion-exchange, size-exclusion, affinity, and high-performance liquid chromatography.
Proteins are separated and purified on the basis of differences in their properties. Proteins can be selectively precipitated by changes in pH or temperature, and particularly by the addition of certain salts. A wide range of chromatographic procedures makes use of differences in size, binding affinities, charge, and other properties. These include ion-exchange, size-exclusion, affinity, and high-performance liquid chromatography.