9.1 Studying Genes and Their Products
A researcher has isolated a new enzyme that she knows is the key to a human disease. She hopes to isolate large amounts of the protein to crystallize it for structural analysis and to study it. She wants to alter amino acid residues at its active site so that she can understand the reaction it catalyzes. She plans an elaborate research program to elucidate how this enzyme interacts with, and is regulated by, other proteins in the cell. All of this, and much more, becomes possible if she can isolate the gene encoding her enzyme. Unfortunately, that gene consists of just a few thousand base pairs within a human chromosome with a size measured in hundreds of millions of base pairs. How does she isolate the small segment that she needs and then study it? The answer lies in DNA cloning and methods developed to manipulate cloned genes.
Genes Can Be Isolated by DNA Cloning
A clone is an identical copy. This term originally applied to cells of a single type, isolated and allowed to reproduce to create a population of identical cells. When applied to DNA, a clone represents many identical copies of a particular gene segment. In brief, our researcher must separate the gene from the larger chromosome, attach it to a much smaller piece of carrier DNA, and allow microorganisms to make many copies of it. This is the process of DNA cloning. The result is selective amplification of a particular gene or DNA segment so that its genetic information may be studied and utilized. Classically, the cloning of DNA from any organism entails five general procedures:
 In brief, our researcher must separate the gene from the larger chromosome, attach it to a much smaller piece of carrier DNA, and allow microorganisms to make many copies of it. This is the process of
In brief, our researcher must separate the gene from the larger chromosome, attach it to a much smaller piece of carrier DNA, and allow microorganisms to make many copies of it. This is the process of - Obtaining the DNA segment to be cloned. Enzymes called restriction endonucleases act as precise molecular scissors, recognizing specific sequences in DNA and cleaving genomic DNA into smaller fragments suitable for cloning. Alternatively, genomic DNA can be sheared randomly into fragments of a desired size. Since the sequence of targeted genomic regions is often known (available in databases), DNA segments to be cloned are most often amplified by the polymerase chain reaction (PCR) or are simply synthesized (both methods are described in Chapter 8).
- Selecting a small molecule of DNA capable of autonomous replication. These small DNAs are called cloning vectors (a vector is a carrier or delivery agent). Most cloning vectors used in the laboratory are modified versions of naturally occurring small DNA molecules found in bacteria or eukaryotes. Viral DNAs may also play this role.
- Joining two DNA fragments covalently. The enzyme DNA ligase links the cloning vector to the DNA fragment to be cloned. Composite DNA molecules of this type, comprising covalently linked segments from two or more sources, are called recombinant DNAs.
- Moving recombinant DNA from the test tube to a host organism. The host organism provides the enzymatic machinery for DNA replication.
- Selecting or identifying host cells that contain recombinant DNA. The cloning vector generally has features that allow the host cells to survive in an environment in which cells lacking the vector would die. Cells containing the vector are thus “selectable” in that environment.
The methods used to accomplish these and related tasks are collectively referred to as recombinant DNA technology or, more informally, genetic engineering.
Much of our initial discussion focuses on DNA cloning in the bacterium Escherichia coli, the first organism used for recombinant DNA work and still the most common host cell. E. coli has many advantages: its DNA metabolism (like many other of its biochemical processes) is well understood; many naturally occurring cloning vectors associated with E. coli, such as plasmids and bacteriophages (bacterial viruses; also called phages), are readily available; and techniques are available for moving DNA expeditiously from one bacterial cell to another. The principles discussed here are broadly applicable to DNA cloning in other organisms, a topic discussed more fully later in the section.
Restriction Endonucleases and DNA Ligases Yield Recombinant DNA
A set of enzymes (Table 9-1) made available through decades of research on nucleic acid metabolism is indispensable for generating and propagating a recombinant DNA molecule (Fig. 9-1). First, restriction endonucleases (also called restriction enzymes) recognize and cleave DNA at specific sequences (recognition sequences or restriction sites) to generate a set of smaller fragments. Second, the DNA fragment to be cloned is joined to a suitable cloning vector by using DNA ligases to link the DNA molecules together. The recombinant vector is then introduced into a host cell, which amplifies the fragment in the course of many generations of cell division.
| Enzyme(s) | Function |
|---|---|
Type II restriction endonucleases |
Cleave DNA molecules at specific base sequences |
DNA ligase |
Joins two DNA molecules or fragments |
DNA polymerase I (E. coli) |
Fills gaps in duplexes by stepwise addition of nucleotides to ends |
Reverse transcriptase |
Makes a DNA copy of an RNA molecule |
Polynucleotide kinase |
Adds a phosphate to the -OH end of a polynucleotide to label it or to permit ligation |
Terminal transferase |
Adds homopolymer tails to the -OH ends of a linear duplex |
Exonuclease III |
Removes nucleotide residues from the ends of a DNA strand |
Bacteriophage λ exonuclease |
Removes nucleotides from the ends of a duplex to expose single-stranded ends |
Alkaline phosphatase |
Removes terminal phosphates from the end or end (or both) |
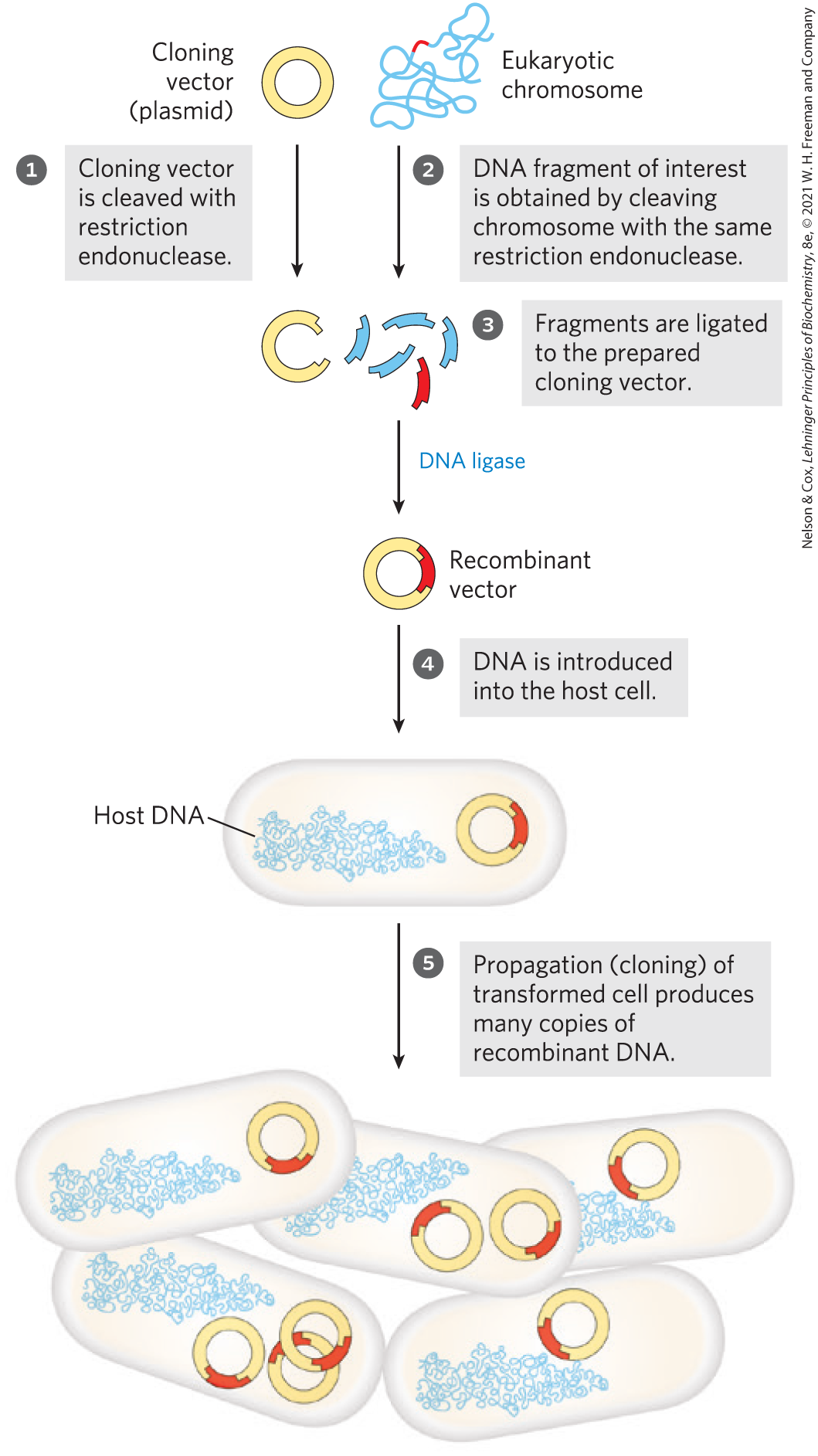
FIGURE 9-1 Schematic illustration of DNA cloning. A cloning vector and eukaryotic chromosomes are separately cleaved with the same restriction endonuclease. (A single chromosome is shown here for simplicity.) The fragments to be cloned are then ligated to the cloning vector. The resulting recombinant DNA (only one recombinant vector is shown here) is introduced into a host cell, where it can be propagated (cloned). Note that this drawing is not to scale: the size of the E. coli chromosome relative to that of a typical cloning vector (such as a plasmid) is much greater than depicted here.
Restriction endonucleases are found in a wide range of bacterial species. As Werner Arber discovered in the early 1960s, the biological function of restriction endonucleases is to recognize and cleave foreign DNA (the DNA of an infecting virus, for example); such DNA is said to be restricted. In the host cell’s DNA, the sequence that would be recognized by one of its own restriction endonucleases is protected from digestion by methylation of the DNA, catalyzed by a specific DNA methylase. The restriction endonuclease and the corresponding methylase are sometimes referred to as a restriction-modification system.
There are three types of restriction endonucleases, designated I, II, and III. Types I and III are generally large, multisubunit complexes containing both the endonuclease and methylase activities. Type II restriction endonucleases, first isolated by Hamilton Smith in 1970, are simpler, require no ATP, and catalyze the hydrolytic cleavage of particular phosphodiester bonds in the DNA within the recognition sequence itself. The extraordinary utility of this group of restriction endonucleases was demonstrated by Daniel Nathans, who first used them to develop novel methods for mapping and analyzing genes and genomes.
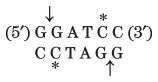
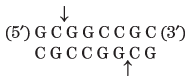
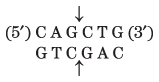
Thousands of type II restriction endonucleases have been discovered in different bacterial species, and more than 100 different DNA sequences are recognized by one or more of these enzymes. The recognition sequences are usually 4 to 6 bp long and are palindromic (see Fig. 8-18). Table 9-2 lists sequences recognized by a few type II restriction endonucleases.
BamHI |
|
HindIII |
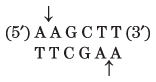
|
ClaI |
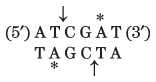
|
NotI |
|
EcoRI |
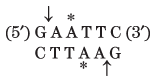
|
PstI |
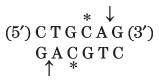
|
EcoRV |

|
PvuII |
|
HaeIII |
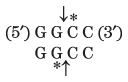
|
Tth111I |

|
Note: Arrows indicate the phosphodiester bonds cleaved by each restriction endonuclease. Asterisks indicate bases that are methylated by the corresponding methylase (where known). N denotes any base. Note that the name of each enzyme consists of a three-letter abbreviation of the bacterial species from which it is derived, sometimes followed by a strain designation and roman numerals to distinguish different restriction endonucleases isolated from the same bacterial species. Thus BamHI is the first (I) restriction endonuclease characterized from Bacillus amyloliquefaciens, strain H. |
|||
Some restriction endonucleases make staggered cuts on the two DNA strands, leaving two to four nucleotides of one strand unpaired at each resulting end. These unpaired strands are referred to as sticky ends (Fig. 9-2a) because they can base-pair with each other or with complementary sticky ends of other DNA fragments. Other restriction endonucleases cleave both strands of DNA straight across, at opposing phosphodiester bonds, leaving no unpaired bases on the ends, often called blunt ends (Fig. 9-2b).
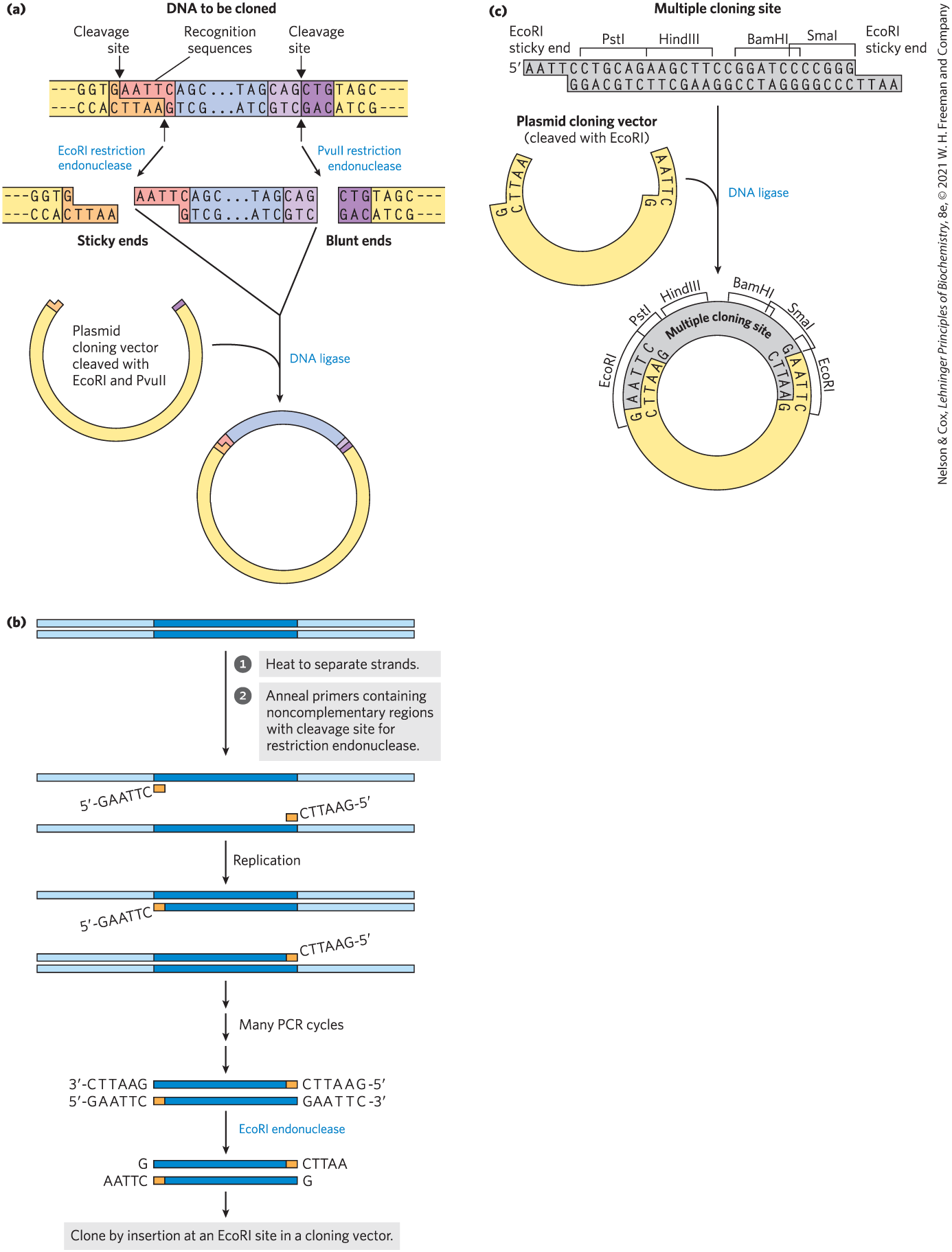
FIGURE 9-2 Use of restriction endonucleases in cloning. (a) Restriction endonucleases recognize and cleave only specific sequences, leaving either sticky ends (with protruding single strands) or blunt ends. Fragments can be ligated to other DNAs, such as the cleaved cloning vector (a plasmid) shown here. This reaction is facilitated by the annealing of complementary sticky ends. Ligation is less efficient for DNA fragments with blunt ends than for those with complementary sticky ends, and DNA fragments with different (noncomplementary) sticky ends generally are not ligated. (b) DNA that has been amplified by the polymerase chain reaction (see Fig. 8-33) can be cloned. The primers can include noncomplementary ends that have a site for cleavage by a restriction endonuclease. Although these parts of the primers do not anneal to the target DNA, the PCR process incorporates them into the DNA that is amplified. Cleavage of the amplified fragments at these sites creates sticky ends, used in ligation of the amplified DNA to a cloning vector. (c) A synthetic DNA fragment with recognition sequences for several restriction endonucleases can be inserted into a plasmid that has been cleaved by a restriction endonuclease. The insert is called a linker; an insert with multiple restriction sites is generally called a multiple cloning site (MCS).
The gene or DNA segment to be cloned is most often generated by the polymerase chain reaction. Careful design of the primers used for PCR (see Fig. 8-33) can alter the amplified segment by the inclusion, at each end, of additional DNA not present in the chromosome that is being targeted. For example, including restriction endonuclease cleavage sites can facilitate the subsequent cloning of the amplified DNA (Fig. 9-2c).
 The gene or DNA segment to be cloned is most often generated by the polymerase chain reaction. Careful design of the primers used for PCR (see
The gene or DNA segment to be cloned is most often generated by the polymerase chain reaction. Careful design of the primers used for PCR (see After the target DNA fragment is prepared and digested with the appropriate restriction enzyme, DNA ligase can be used to join it to a vector digested by the same restriction endonuclease; a fragment generated by EcoRI, for example, generally will not link to a fragment generated by BamHI. As described in more detail in Chapter 25 (see Fig. 25-15), DNA ligase catalyzes the formation of new phosphodiester bonds in a reaction that uses ATP or a similar cofactor. The base pairing of complementary sticky ends greatly facilitates the ligation reaction (Fig. 9-2a). Blunt ends can also be ligated, albeit less efficiently. Researchers can create new DNA sequences for a wide range of purposes by inserting synthetic DNA fragments, called linkers, to bridge the ends that are being ligated. An inserted DNA fragment with multiple recognition sequences for restriction endonucleases (often useful later as points for inserting additional DNA by cleavage and ligation) is called the multiple cloning site (MCS) (Fig. 9-2d).
 After the target DNA fragment is prepared and digested with the appropriate restriction enzyme, DNA ligase can be used to join it to a vector digested by the same restriction endonuclease; a fragment generated by EcoRI, for example, generally will not link to a fragment generated by BamHI. As described in more detail in
After the target DNA fragment is prepared and digested with the appropriate restriction enzyme, DNA ligase can be used to join it to a vector digested by the same restriction endonuclease; a fragment generated by EcoRI, for example, generally will not link to a fragment generated by BamHI. As described in more detail in The effectiveness of sticky ends in selectively joining two DNA fragments was apparent in the earliest recombinant DNA experiments. Before restriction endonucleases were widely available, some investigators found they could generate sticky ends by the combined action of the bacteriophage λ exonuclease and terminal transferase (Table 9-1). The fragments to be joined were given complementary homopolymeric tails. Peter Lobban and Dale Kaiser used this method in 1971 in the first experiments to join naturally occurring DNA fragments. Similar methods were used soon after in Paul Berg’s laboratory to join DNA segments from simian virus 40 (SV40) to DNA derived from bacteriophage λ, thereby creating the first recombinant DNA molecule with DNA segments from different species.
Cloning Vectors Allow Amplification of Inserted DNA Segments
The factors that govern the delivery of recombinant DNA in clonable form to a host cell, and its subsequent amplification in the host, are well illustrated in three popular cloning vectors: plasmids and bacterial artificial chromosomes, used in experiments with E. coli, and a vector used to clone large DNA segments in yeast.
Plasmids
A plasmid is a circular DNA molecule that replicates separately from the host chromosome. Naturally occurring bacterial plasmids range in size from 5,000 to 400,000 bp. Many of the plasmids found in bacterial populations are little more than molecular parasites, similar to viruses but with a more limited capacity to transfer from one cell to another. To survive in the host cell, plasmids incorporate several specialized sequences that enable them to make use of the cell’s resources for their own replication and gene expression.
Naturally occurring plasmids usually have a symbiotic role in the cell. They may provide genes that confer resistance to antibiotics or that perform new functions for the cell. For example, the Ti plasmid of Agrobacterium tumefaciens allows the host bacterium to colonize the cells of a plant and make use of the plant’s resources. The same properties that enable plasmids to grow and survive in a bacterial or eukaryotic host are useful to molecular biologists who want to engineer a vector for cloning a specific DNA segment. Constructed in 1977, one of the first recombinant vectors — E. coli plasmid pBR322 — illustrates some key features that define a useful cloning vector (Fig. 9-3):
- The plasmid pBR322 has an origin of replication, or ori, a sequence where replication is initiated by cellular enzymes (see Chapter 25). This sequence is required to propagate the plasmid. An associated regulatory system is present that limits replication to maintain pBR322 at a level of 10 to 20 copies per cell.
- The plasmid contains genes that confer resistance to the antibiotics ampicillin () and tetracycline (), allowing the selection of cells that contain the intact plasmid or a recombinant version of the plasmid (discussed below).
- Several unique recognition sequences in pBR322 are targets for restriction endonucleases (PstI, EcoRI, BamHI, SalI, and PvuII), providing sites where the plasmid can be cut to insert foreign DNA.
- The small size of the plasmid (4,361 bp) facilitates its entry into cells and the biochemical manipulation of the DNA. This small size was the result of trimming away many DNA segments from a larger, parent plasmid — sequences that the biochemist does not need.
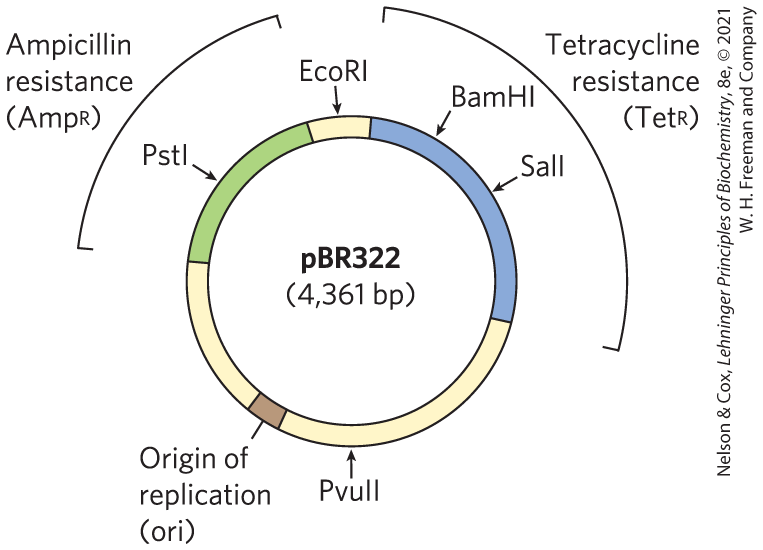
FIGURE 9-3 The constructed E. coli plasmid pBR322. Notice the location of some important restriction sites, for PstI, EcoRI, BamHI, SalI, and PvuII; genes for ampicillin and tetracycline resistance ( and ); and the replication origin (ori). Constructed in 1977, this was one of the early plasmids designed expressly for cloning in E. coli.
The replication origins inserted in common plasmid vectors were originally derived from naturally occurring plasmids. As in pBR322, each of these origins is regulated to maintain a particular plasmid copy number. Depending on the origin used, the plasmid copy number can vary from one to hundreds or thousands per cell, providing many options for investigators. Two different plasmids cannot function in the same cell if they use the same origin of replication, because the regulation of one will interfere with the replication of the other. Such plasmids are said to be incompatible. When a researcher wants to introduce two or more different plasmids into a bacterial cell, each plasmid must have a different replication origin.
In the laboratory, small plasmids can be introduced into bacterial cells by a process called transformation. The cells (often E. coli, but other bacterial species are also used) and plasmid DNA are incubated together at in a calcium chloride solution, then are subjected to heat shock by rapidly shifting the temperature to between and . For reasons not well understood, some of the cells treated in this way take up the plasmid DNA. Some species of bacteria, such as Acinetobacter baylyi, are naturally competent for DNA uptake and do not require the calcium chloride–heat shock treatment. In an alternative method, called electroporation, cells incubated with the plasmid DNA are subjected to a high-voltage pulse, which transiently renders the bacterial membrane permeable to large molecules.
Regardless of the approach, relatively few cells take up the plasmid DNA, so a method is needed to identify those that do. The usual strategy is to utilize one of two types of genes in the plasmid, referred to as selectable and screenable markers. A selectable marker either permits the growth of a cell (positive selection) or kills the cell (negative selection) under a defined set of conditions. The plasmid pBR322 provides markers for both positive and negative selection (Fig. 9-4). A screenable marker is a gene encoding a protein that causes the cell to produce a colored or fluorescent molecule. Cells are not harmed when the gene is present, and the cells that carry the plasmid are easily identified by the colored or fluorescent colonies they produce.
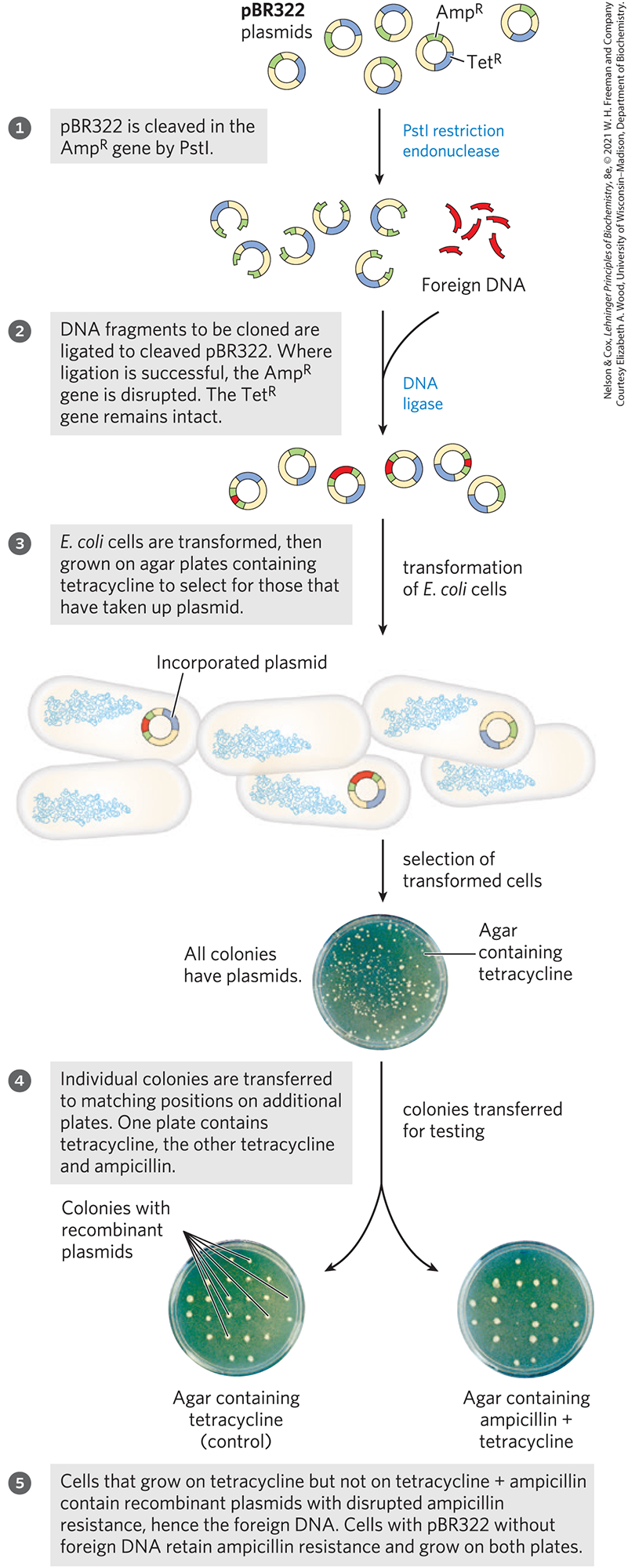
FIGURE 9-4 Use of pBR322 to clone foreign DNA in E. coli and identify cells containing the DNA.
Transformation of typical bacterial cells with purified DNA (never a very efficient process) becomes less successful as plasmid size increases, and it is difficult to clone DNA segments longer than about 15,000 bp when plasmids are used as the vector.
To illustrate the use of a plasmid as a cloning vector, consider the bacterial gene encoding a recombinase called the RecA protein (see Chapter 25). In most bacteria, the gene encoding RecA is one of the thousands of genes on a chromosome millions of base pairs long. The recA gene is just over 1,000 bp long. A plasmid would be a good choice for cloning a gene of this size. As described later, the cloned gene can be altered in a variety of ways, and the gene variants can be expressed at high levels to enable purification of the encoded protein.
Bacterial Artificial Chromosomes
Researchers sometimes want to clone much longer DNA segments than can typically be incorporated into standard plasmid cloning vectors such as pBR322. To meet this need, plasmid vectors have been developed with special features that allow the cloning of very long segments (typically 100,000 to 300,000 bp) of DNA. Once such large segments of cloned DNA have been added, these vectors are large enough to be thought of as chromosomes and are known as bacterial artificial chromosomes, or BACs (Fig. 9-5).
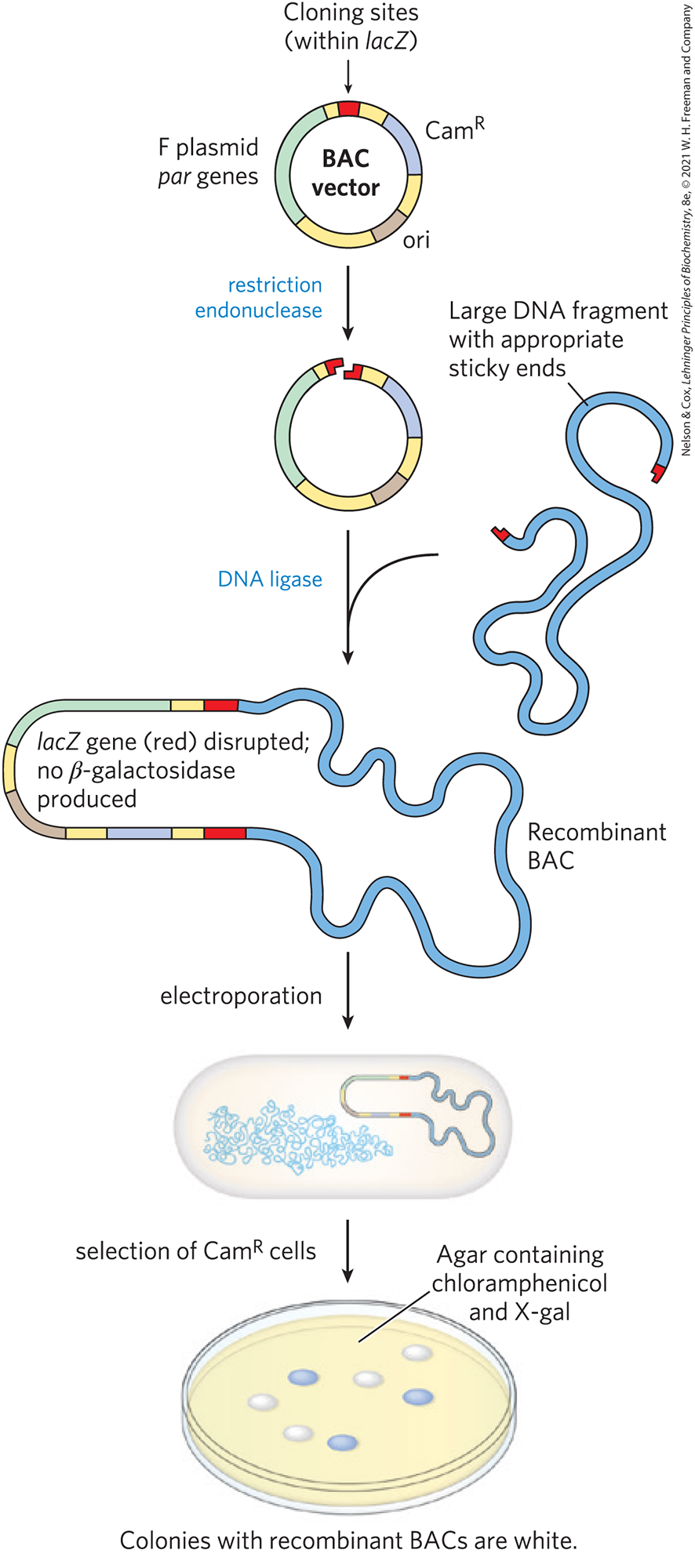
FIGURE 9-5 Bacterial artificial chromosomes (BACs) as cloning vectors. The vector is a relatively simple plasmid, with a replication origin (ori) that directs replication. The par genes assist in the even distribution of plasmids to daughter cells at cell division. This increases the likelihood of each daughter cell carrying one copy of the plasmid, even when few copies are present. The low number of copies is useful in cloning large segments of DNA, because this limits the opportunities for unwanted recombination reactions that can unpredictably alter large cloned DNAs over time. The BAC includes selectable markers. A lacZ gene (required for the production of the enzyme β-galactosidase) is situated in the cloning region such that it is inactivated by cloned DNA inserts. Introduction of recombinant BACs into cells by electroporation is promoted by the use of cells with an altered (more porous) cell wall. Recombinant DNAs are screened for resistance to the antibiotic chloramphenicol (). Plates also contain X-gal, a substrate for β-galactosidase that yields a blue product. Colonies with active β-galactosidase, and hence no DNA insert in the BAC vector, turn blue; colonies without β-galactosidase activity, and thus with the desired DNA inserts, are white.
A BAC vector (without any cloned DNA inserted) is a relatively simple plasmid, generally not much larger than other plasmid vectors. To accommodate very long segments of cloned DNA, BAC vectors have stable origins of replication that maintain the plasmid at one or two copies per cell. The low copy number is useful in cloning large segments of DNA, because it limits the opportunities for unwanted recombination reactions that can unpredictably alter large cloned DNAs over time. BACs also include par genes, derived from a type of plasmid called an F plasmid. The par genes encode proteins that direct the reliable distribution of the recombinant chromosomes to daughter cells at cell division, thereby increasing the likelihood of each daughter cell carrying one copy, even when few copies are present.
The BAC vector includes both selectable and screenable markers. The BAC vector shown in Figure 9-5 contains a gene that confers resistance to the antibiotic chloramphenicol (). Vector-containing cells can be selected by growing them on agar plates containing this antibiotic — a positive selection, as the cells with the vector survive. A lacZ gene, required for production of the enzyme β-galactosidase, is a screenable marker that can reveal which cells contain plasmids — now chromosomes — that incorporate the cloned DNA segments. The β-galactosidase catalyzes conversion of the colorless molecule 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (more simply, X-gal) to a blue product. If the gene is intact and expressed, the colony containing it is blue. If gene expression is disrupted by the introduction of a cloned DNA segment, the colony is white.
Yeast Artificial Chromosomes
As with E. coli, yeast genetics is a well-developed discipline. Research on large genomes and the associated need for high-capacity cloning vectors led to the development of yeast artificial chromosomes, or YACs (Fig. 9-6). As with BACs, YAC vectors can be used to clone very long segments of DNA. In addition, the DNA cloned in a YAC can be altered to study the function of specialized sequences in chromosome metabolism, mechanisms of gene regulation and expression, and many other aspects of eukaryotic molecular biology.
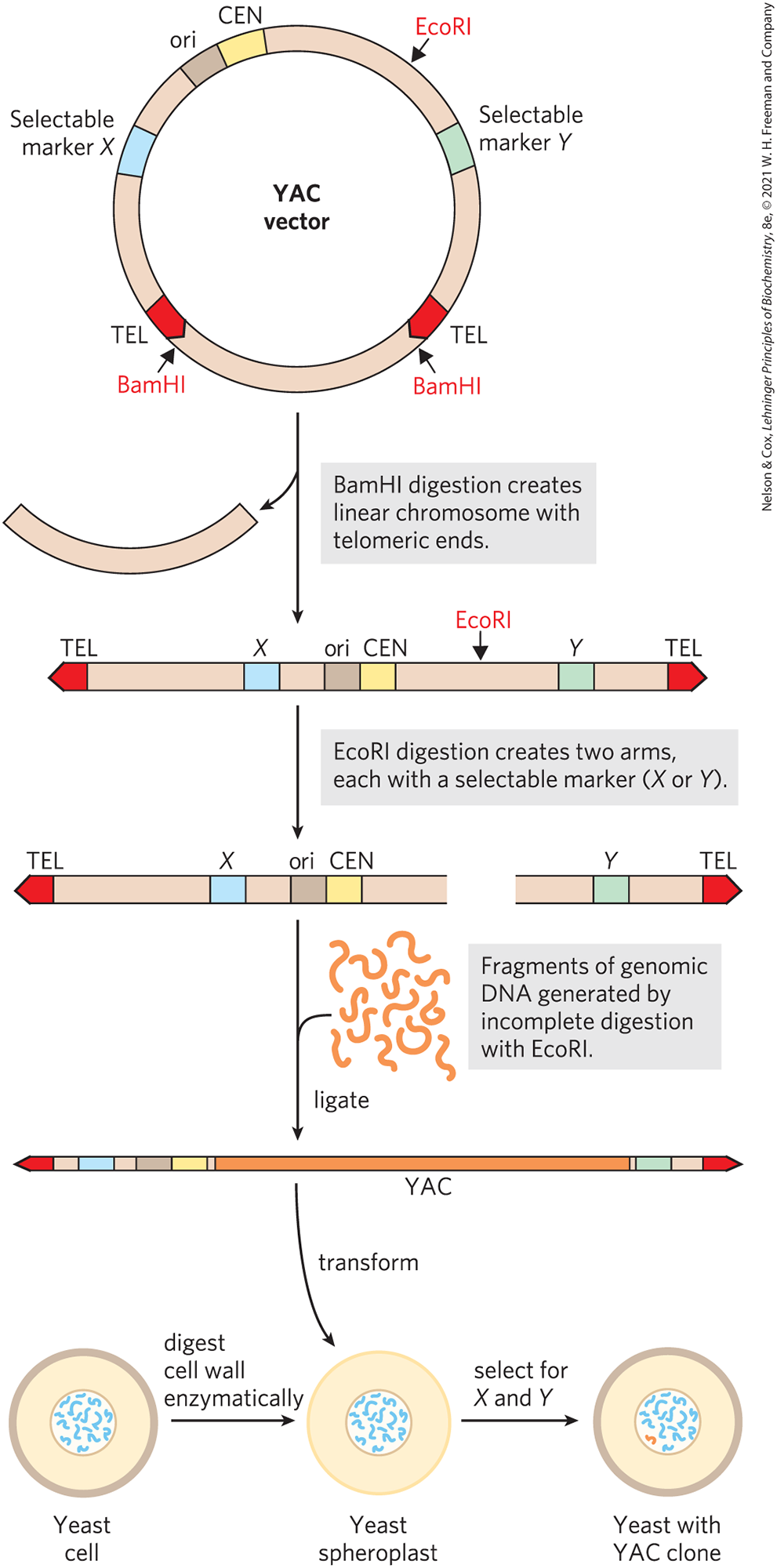
FIGURE 9-6 Construction of a yeast artificial chromosome (YAC). A YAC vector includes an origin of replication (ori), a centromere (CEN), two telomeres (TEL), and selectable markers (X and Y). Digestion with BamHI and EcoRI generates two separate DNA arms, each with a telomeric end and one selectable marker. A large segment of DNA (e.g., up to bp from the human genome) is ligated to the two arms to create a yeast artificial chromosome. The YAC transforms yeast cells (prepared by removal of the cell wall to form spheroplasts), and the cells are selected for X and Y; the surviving cells propagate the DNA insert.
The genome of Saccharomyces cerevisiae contains only (less than four times the size of the E. coli chromosome), and its entire sequence is known. Yeast is also very easy to maintain and grow on a large scale in the laboratory. Plasmid vectors have been constructed for insertions into yeast cells, employing the same principles that govern the use of E. coli vectors. Convenient methods for moving DNA into and out of yeast cells permit the study of many aspects of eukaryotic cell biochemistry. Some recombinant plasmids incorporate multiple replication origins and other elements that allow them to be used in more than one species (e.g., in yeast and in E. coli). Plasmids that can be propagated in cells of two or more species are called shuttle vectors.
YAC vectors contain all the elements needed to maintain a eukaryotic chromosome in the yeast nucleus: a yeast origin of replication, two selectable markers, and specialized sequences (derived from the telomeres and centromere) needed for stability and proper segregation of the chromosomes at cell division (see Chapter 24). In preparation for its use in cloning, the vector is propagated as a circular bacterial plasmid and then isolated and purified. Cleavage with a restriction endonuclease (BamHI in Fig. 9-6) removes a length of DNA between two telomere sequences (TEL), leaving the telomeres at the ends of the linearized DNA. Cleavage at another internal site (by EcoRI in Fig. 9-6) divides the vector into two DNA segments, referred to as vector arms, each with a different selectable marker.
Genomic DNA to be cloned is prepared by partial digestion with restriction endonucleases to obtain a suitable fragment size. Genomic fragments are then separated by pulsed field gel electrophoresis, a variation of gel electrophoresis (see Fig. 3-18) that segregates very large DNA segments. DNA fragments of appropriate size (up to about ) are mixed with the prepared vector arms and ligated. The ligation mixture is then used to transform yeast cells (pretreated to partially degrade their cell walls) with these very large DNA molecules — which now have the structure and size to be considered yeast chromosomes. Culture on a medium that requires the presence of both selectable marker genes ensures the growth of only those yeast cells that contain an artificial chromosome with a large insert sandwiched between the two vector arms (Fig. 9-6). The stability of YAC clones increases with the length of the cloned DNA segment (up to a point). Those with inserts of more than 150,000 bp are nearly as stable as normal cellular chromosomes, whereas those with inserts of fewer than 100,000 bp are gradually lost during mitosis (so, generally, there are no yeast cell clones carrying only the two vector ends ligated together or vectors with only short inserts). YACs that lack a telomere at either end are rapidly degraded.
Cloned Genes Can Be Expressed to Amplify Protein Production
Frequently, the product of a cloned gene, rather than the gene itself, is of primary interest — particularly when the protein has commercial, therapeutic, or research value. Proteins are encoded by genes in DNA; alter the DNA in a gene, and one can alter the protein product of that gene. Biochemists use purified proteins for many purposes, including to elucidate protein function, study reaction mechanisms, generate antibodies to the proteins, reconstitute complex cellular activities in the test tube with purified components, and examine protein binding partners. With an increased understanding of the fundamentals of DNA, RNA, and protein metabolism and their regulation in a host organism such as E. coli or yeast, investigators can manipulate cells to express cloned genes in order to study their protein products. The general goal is to alter the sequences around a cloned gene to trick the host organism into producing the protein product of the gene, often at very high levels. This overexpression of a protein can make its subsequent purification much easier.
We’ll use the expression of a eukaryotic protein in a bacterium as an example. Eukaryotic genes have surrounding sequences needed for their transcription and regulation in the cells they are derived from, but these sequences do not function in bacteria. Thus, eukaryotic genes lack the DNA sequence elements required for their controlled expression in bacterial cells: promoters (sequences that instruct RNA polymerase where to bind to initiate mRNA synthesis), ribosome-binding sites (sequences that allow translation of the mRNA to protein), and additional regulatory sequences. Appropriate bacterial regulatory sequences for transcription and translation must be inserted in the vector DNA at the correct positions relative to the eukaryotic gene.
Cloning vectors with the transcription and translation signals needed for the regulated expression of a cloned gene are called expression vectors. The rate of expression of the cloned gene is controlled by replacing the gene’s normal promoter and regulatory sequences with more efficient and convenient versions supplied by the vector. Generally, a well-characterized promoter and its regulatory elements are positioned near several unique restriction sites for cloning, so that genes inserted at the restriction sites will be expressed from the regulated promoter elements (Fig. 9-7). Some of these vectors incorporate other features, such as a bacterial ribosome-binding site to enhance translation of the mRNA derived from the gene (Chapter 27) or a transcription termination sequence (Chapter 26). In some cases, cloned genes are so efficiently expressed that their protein product represents 10% or more of the cellular protein. At these concentrations, some foreign proteins can kill the host cell (usually E. coli), so expression of the cloned gene must be limited to the few hours before the planned harvesting of the cells.
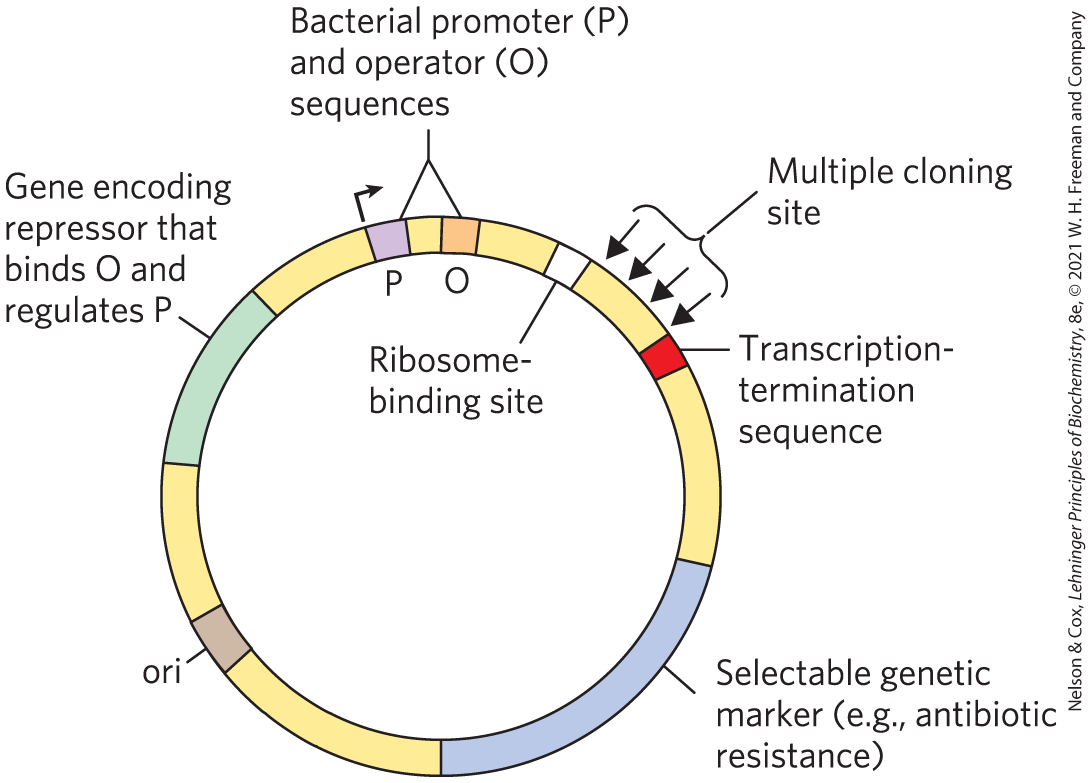
FIGURE 9-7 DNA sequences in a typical E. coli expression vector. The gene to be expressed is inserted into one of the restriction sites in the MCS, near the promoter (P), with the end of the gene encoding the amino terminus of the protein positioned closest to the promoter. The promoter allows efficient transcription of the inserted gene, and the transcription-termination sequence sometimes improves the amount and stability of the mRNA produced. The operator (O) permits regulation by a repressor that binds to it. The ribosome-binding site provides sequence signals for the efficient translation of the mRNA derived from the gene. The selectable marker allows the selection of cells containing the recombinant DNA.
Many Different Systems Are Used to Express Recombinant Proteins
Every living organism has the capacity to express genes in its genomic DNA; thus, in principle, any organism can serve as a host to express proteins from a different (heterologous) species. Almost every sort of organism has, indeed, been used for this purpose, and each host type has a particular set of advantages and disadvantages.
Bacteria
Bacteria, especially E. coli, remain the most common hosts for protein expression. The regulatory sequences that govern gene expression in E. coli and many other bacteria are well understood and can be harnessed to express cloned proteins at high levels. Bacteria are easy to store and grow in the laboratory, on inexpensive growth media. Efficient methods also exist to get DNA into bacteria and extract DNA from them. Bacteria can be grown in huge amounts in commercial fermenters, providing a rich source of the cloned protein.
Problems do exist, however. When expressed in bacteria, some heterologous proteins do not fold correctly, and many do not undergo the posttranslational modifications or proteolytic cleavage that may be necessary for their activity. Certain features of a gene sequence also can make a particular gene difficult to express in bacteria. For example, intrinsically disordered regions are more common in eukaryotic proteins. When expressed in bacteria, many eukaryotic proteins aggregate into insoluble cellular precipitates called inclusion bodies. For these and many other reasons, some eukaryotic proteins are inactive when purified from bacteria or cannot be expressed at all. To help address some of these problems, researchers are regularly developing new bacterial host strains that include enhancements such as the engineered presence of eukaryotic protein chaperones or enzymes that modify eukaryotic proteins.
There are many specialized systems for expressing proteins in bacteria. The well-characterized promoter and regulatory sequences associated with the lactose operon (see Chapter 28) are often fused to the gene of interest to direct transcription. The cloned gene will be transcribed when lactose is added to the growth medium. However, regulation in the lactose system is “leaky”: it is not turned off completely when lactose is absent — a potential problem if the product of the cloned gene is toxic to the host cells. Transcription from the Lac promoter is also not efficient enough for some applications.
An alternative system uses the promoter and RNA polymerase of a bacterial virus called bacteriophage T7. If the cloned gene is fused to a T7 promoter, it is transcribed, not by the E. coli RNA polymerase, but by the T7 RNA polymerase. The gene encoding this polymerase is separately cloned into the same cell in a construct that affords tight regulation (allowing controlled production of the T7 RNA polymerase). The polymerase is also very efficient and directs high levels of expression of most genes fused to the T7 promoter. This system has been used to express the RecA protein in bacterial cells (Fig. 9-8).
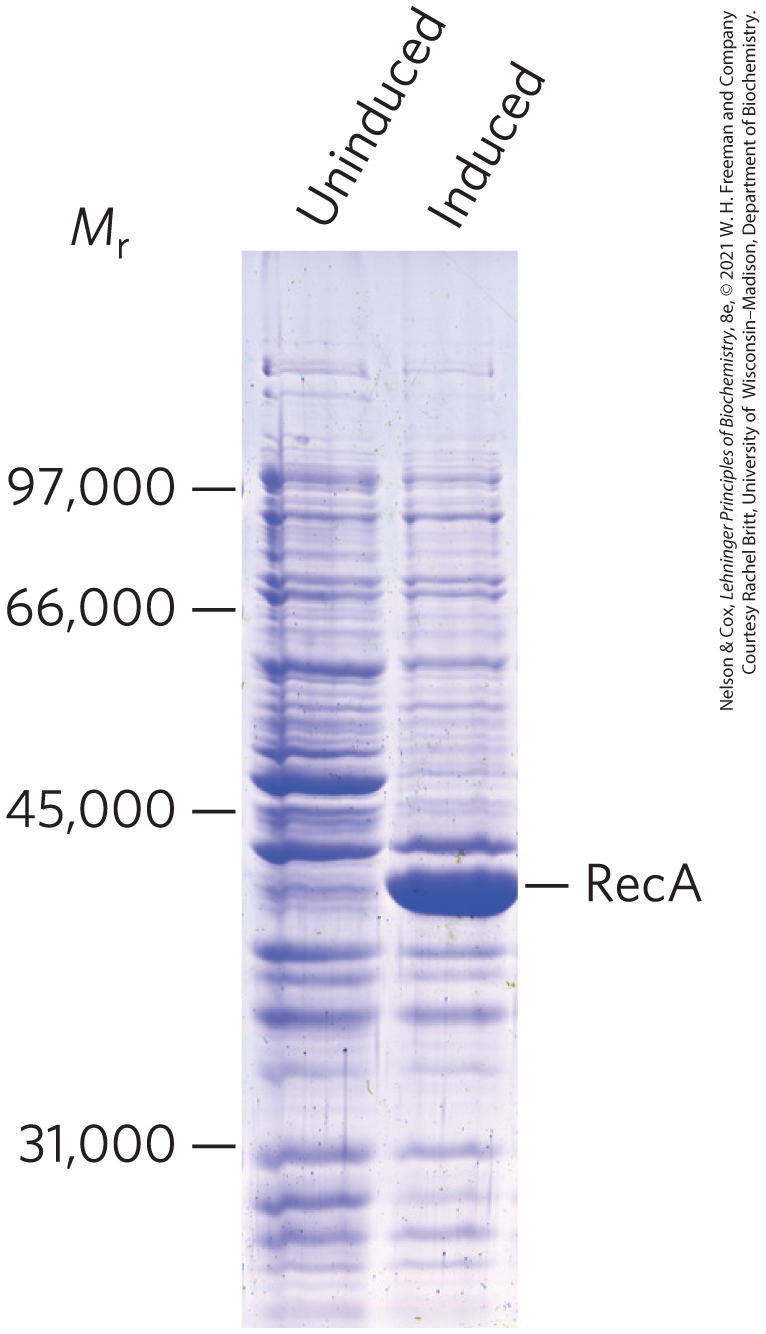
FIGURE 9-8 Regulated expression of RecA protein in a bacterial cell. The gene encoding the RecA protein, fused to a bacteriophage T7 promoter, is cloned into an expression vector. Under normal growth conditions (uninduced), no RecA protein appears. When the T7 RNA polymerase is induced in the cell, the recA gene is expressed, and large amounts of RecA protein are produced. The positions of standard molecular weight markers that were run on the same gel are indicated.
Yeast
Saccharomyces cerevisiae is probably the best understood eukaryotic organism. The principles underlying the expression of a protein in yeast are the same as those for bacteria. Cloned genes must be linked to promoters that can direct high-level expression in yeast cells. For example, the yeast GAL1 and GAL10 genes (encoding enzymes involved in galactose metabolism) are under cellular regulation such that they are expressed when yeast cells are grown in media with galactose but shut down when the cells are grown in glucose. Thus, if a heterologous gene is expressed using these same regulatory sequences, the expression of that gene can be controlled simply by choosing an appropriate medium for cell growth.
Some of the same problems that accompany protein expression in bacteria also occur with yeast. Heterologous proteins may not fold properly, yeast may lack the enzymes needed to modify the proteins to their active forms, or certain features of the gene sequence may hinder expression of a protein. However, because S. cerevisiae is a eukaryote, the expression of eukaryotic genes (especially yeast genes) is sometimes more efficient in this host than in bacteria. As yeast possess many of the same protein chaperones and modification systems of higher eukaryotes, protein products may also be folded and modified more accurately than are proteins expressed in bacteria.
Insects and Insect Viruses
Baculoviruses are insect viruses with double-stranded DNA genomes. When baculoviruses infect their insect larval hosts, they act as parasites, killing the larvae and turning them into factories for virus production. Late in the infection process, the viruses produce large amounts of two proteins (p10 and polyhedrin), neither of which is needed for production of viruses in cultured insect cells. The genes for both of these proteins can be replaced with the gene for a heterologous protein. When the resulting recombinant virus is used to infect insect cells or larvae, the heterologous protein is often produced at very high levels — up to 25% of the total protein present at the end of the infection cycle.
Autographa californica multicapsid nucleopolyhedrovirus (AcMNPV; A. californica is a moth species that it infects) is the baculovirus most often used for protein expression. It has a large genome (134,000 bp), too large for direct cloning. Virus purification is also cumbersome. These problems have been solved by the creation of bacmids, large circular DNAs that include the entire baculovirus genome along with sequences that allow replication of the bacmid in E. coli (Fig. 9-9). The gene of interest is cloned into a smaller plasmid and combined with the larger plasmid by site-specific recombination in vivo (see Fig. 25-37). The recombinant bacmid is then isolated and transfected into insect cells (the term transfection is used when the DNA used for transformation includes viral sequences and leads to viral replication), followed by recovery of the protein once the infection cycle is finished. A wide range of bacmid systems are available commercially. Baculovirus systems are not successful with all proteins. However, with these systems, insect cells sometimes successfully replicate the protein-modification patterns of higher eukaryotes and produce active, correctly modified eukaryotic proteins.
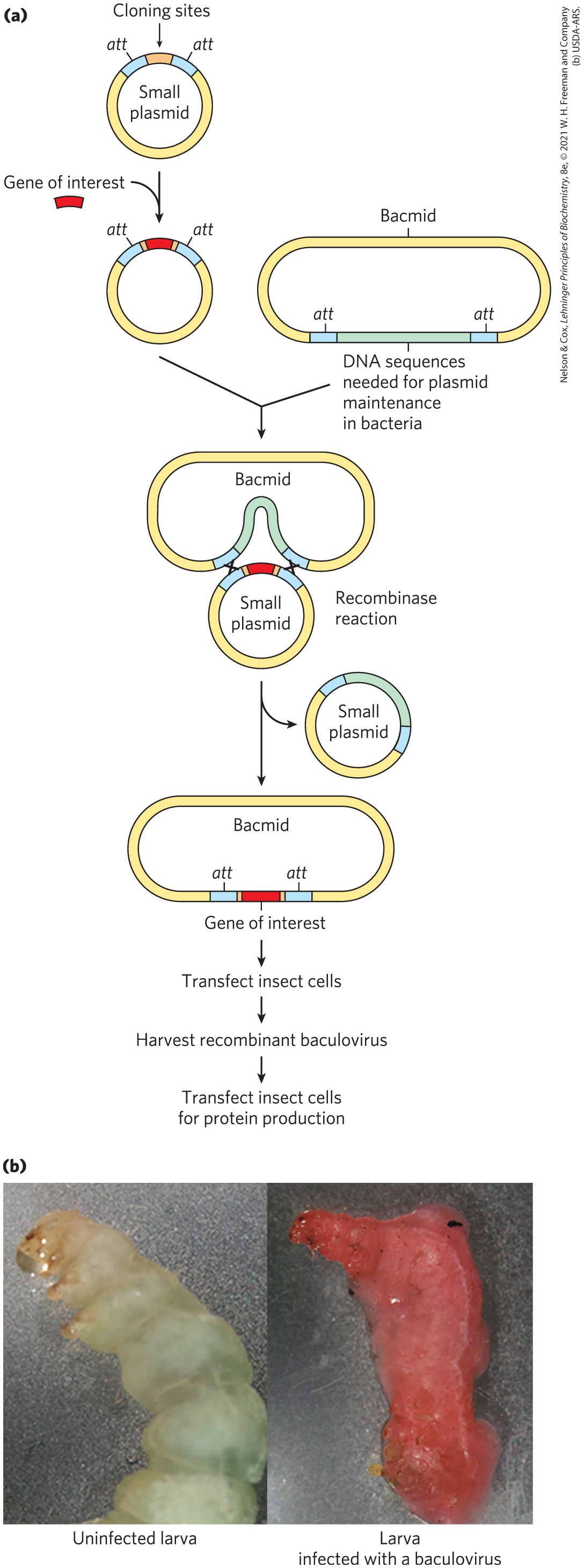
FIGURE 9-9 Cloning with baculoviruses. (a) Shown here is the construction of a typical vector used for protein expression in baculoviruses. The gene of interest is cloned into a small plasmid (top left) between two sites (att) recognized by a site-specific recombinase, then is introduced into the baculovirus vector by site-specific recombination. This generates a circular DNA product that is used to infect the cells of an insect larva. The gene of interest is expressed during the infection cycle, downstream of a promoter that normally expresses a baculovirus coat protein at very high levels. (b) The photographs show larvae of the cabbage looper moth. The larva on the left is uninfected; the larva on the right was infected with a recombinant baculovirus vector expressing a protein that produces a red color.
Mammalian Cells in Culture
The most convenient way to introduce cloned genes into a mammalian cell is with viruses. This method takes advantage of the natural capacity of a virus to insert its DNA or RNA into a cell, and sometimes into the cellular chromosome. A variety of engineered mammalian viruses are available as vectors, including human adenoviruses and retroviruses. The gene of interest is cloned so that its expression is controlled by a virus promoter. The virus uses its natural infection mechanisms to introduce the recombinant genome into cells, where the cloned protein is expressed. One advantage of these systems is that proteins can be expressed either transiently (if the viral DNA is maintained separately from the host cell genome and eventually degraded) or permanently (if the viral DNA is integrated into the host cell genome). With the correct choice of host cell, the proper posttranslational modification of the protein to its active form can be ensured. However, the growth of mammalian cells in tissue culture is very expensive, and this technology is generally used to test the function of a protein in vivo rather than to produce a protein in large amounts.
Alteration of Cloned Genes Produces Altered Proteins
Cloning techniques can be used not only to overproduce proteins but also to produce proteins that are altered, subtly or dramatically, from their native forms. Specific amino acids may be replaced individually by site-directed mutagenesis. This approach has greatly enhanced research on proteins by allowing investigators to make specific changes in the primary structure and examine the effects of these changes on the protein’s folding, three-dimensional structure, and activity. The amino acid sequence of the protein is changed by altering the DNA sequence of the cloned gene. If appropriate restriction sites flank the sequence to be altered, researchers can simply remove a DNA segment and replace it with a synthetic one, identical to the original except for the desired change (Fig. 9-10a).
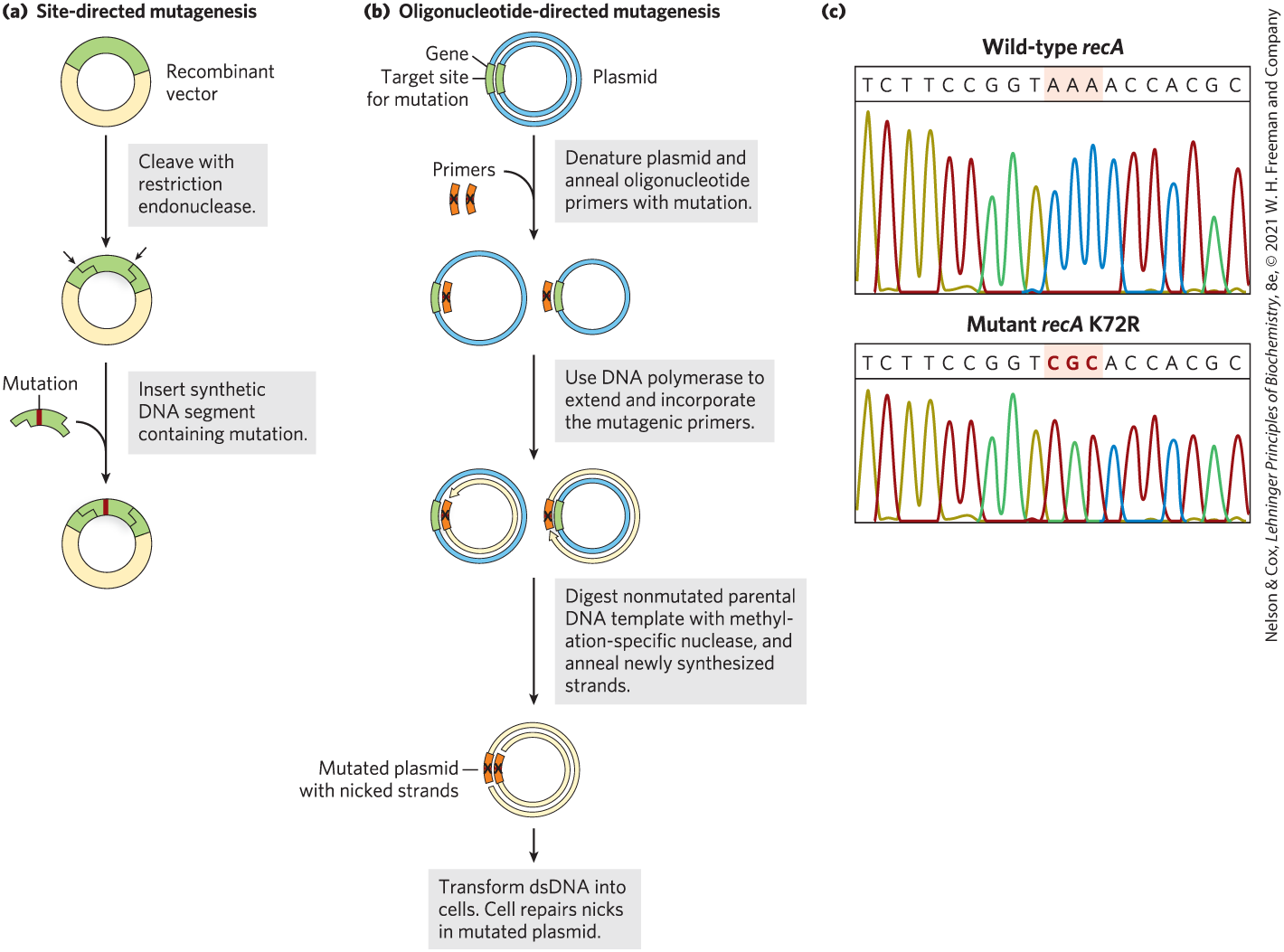
FIGURE 9-10 Two approaches to site-directed mutagenesis. (a) A synthetic DNA segment replaces a fragment removed by a restriction endonuclease. (b) A pair of synthetic and complementary oligonucleotides with a specific sequence change at one position are hybridized to a circular plasmid with a cloned copy of the gene to be altered. The mutated oligonucleotides act as primers for the synthesis of full-length double-stranded (ds) DNA copies of the plasmid that contain the specified sequence change. The blue parental strand was methylated while replicating in its host cell, prior to plasmid isolation. These plasmid copies are then used to transform cells. (c) Results from an automated sequencer (see Fig. 8-35), showing sequences from the wild-type recA gene (top) and an altered recA gene (bottom), with the triplet (codon) at position 72 changed from AAA to CGC, specifying an Arg (R) residue instead of a Lys (K) residue. [(c) Information from Elizabeth A. Wood, University of Wisconsin–Madison, Department of Biochemistry.]
When suitably located restriction sites are not present, oligonucleotide-directed mutagenesis can create a specific DNA sequence change (Fig. 9-10b). The cloned gene is denatured, separating the strands. Two short, complementary synthetic DNA strands, each with the desired base change, are annealed to opposite strands of the cloned gene within a suitable circular DNA vector. The mismatch of a single base pair in 30 to 40 bp does not prevent annealing. The two annealed oligonucleotides serve to prime DNA synthesis in both directions around the plasmid vector, creating two complementary strands that contain the mutation. After several cycles of selective amplification by the polymerase chain reaction (PCR; see Fig. 8-33), the mutation-containing DNA predominates in the population and can be used to transform bacteria. Most of the transformed bacteria will have plasmids carrying the mutation.
For an example, we go back to the bacterial recA gene. The product of this gene, the RecA protein, has several activities (see Section 25.3) including the hydrolysis of ATP. The Lys residue at position 72 in RecA (a 352 residue polypeptide) is involved in ATP hydrolysis. Changing to an Arg creates a variant of RecA protein that will bind, but not hydrolyze, ATP (Fig. 9-10c). The engineering and purification of this variant RecA protein has facilitated research into the roles of ATP hydrolysis in the functioning of this protein.
Changes can be introduced into a gene that involve far more than one base pair. Large parts of a gene can be deleted by cutting out a segment with restriction endonucleases and ligating the remaining portions to form a smaller gene. For example, if a protein has two domains, the gene segment encoding one of the domains can be removed so that the gene now encodes a protein with only one of the original two domains. Parts of two different genes can be ligated to create new combinations; the product of such a fused gene is called a fusion protein. Researchers have ingenious methods to bring about virtually any genetic alteration in vitro. After reintroducing the altered DNA into the cell, they can investigate the consequences of the alteration.
Terminal Tags Provide Handles for Affinity Purification
Affinity chromatography is one of the most efficient methods for purifying proteins (see Fig. 3-17c). Unfortunately, many proteins do not bind a ligand that can be conveniently immobilized on a column matrix. However, the gene for almost any protein can be altered to express a fusion protein that can be purified by affinity chromatography. The gene encoding the target protein is fused to a gene encoding a peptide or protein that binds a simple, stable ligand with high affinity and specificity. The peptide or protein used for this purpose is referred to as a tag. Tag sequences can be added to genes such that the resulting proteins have tags at their amino terminus or carboxyl terminus. Table 9-3 lists some of the peptides or proteins commonly used as tags.
| Tag protein/peptide | Molecular mass (kDa) | Immobilized ligand |
|---|---|---|
Protein A |
59 |
Fc portion of IgG |
0.8 |
||
Glutathione-S-transferase (GST) |
26 |
Glutathione |
Maltose-binding protein |
41 |
Maltose |
β-Galactosidase |
116 |
p-Aminophenyl-β-d-thiogalactoside (TPEG) |
Chitin-binding domain |
5.7 |
Chitin |
The general procedure can be illustrated by focusing on a system that uses the glutathione-S-transferase (GST) tag (Fig. 9-11). GST is a small enzyme ( 26,000) that binds tightly and specifically to glutathione. When the GST gene sequence is fused to a target gene, the fusion protein acquires the capacity to bind glutathione. The fusion protein is expressed in a host organism such as a bacterium, and a crude extract is prepared. A column is filled with a porous matrix consisting of the ligand (glutathione) immobilized on microscopic beads of a stable polymer such as cross-linked agarose. As the crude extract percolates through this matrix, the fusion protein becomes immobilized by binding the glutathione. The other proteins in the extract are washed through the column and discarded. The interaction between GST and glutathione is tight but noncovalent, allowing the fusion protein to be gently eluted from the column with a solution containing either a higher concentration of salts or free glutathione to compete with the immobilized ligand for GST binding. The fusion protein is often obtained with good yield and high purity. In some commercially available systems, the tag can be entirely or largely removed from the purified fusion protein by a protease that cleaves a sequence near the junction between the target protein and its tag.
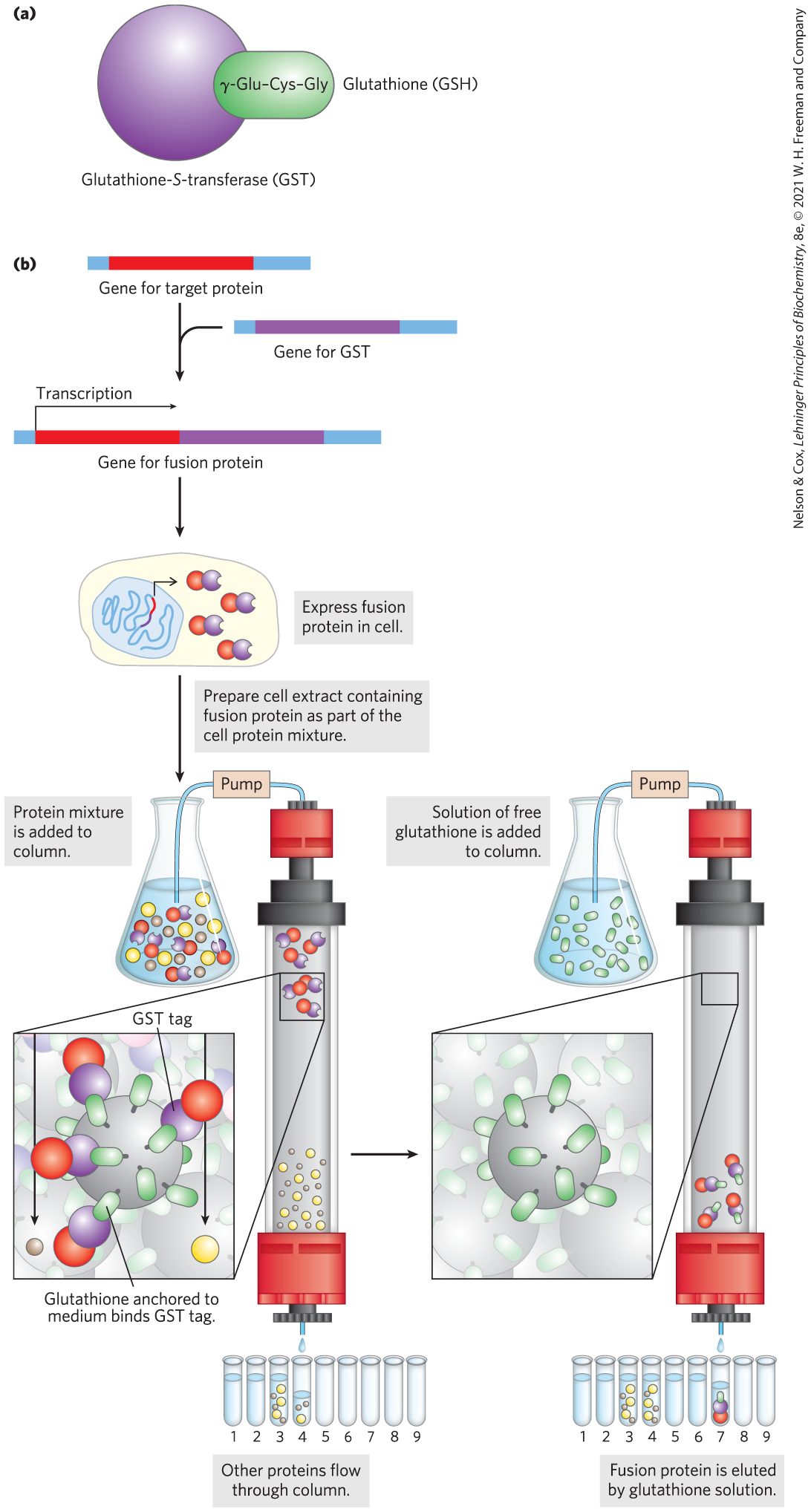
FIGURE 9-11 Use of tagged proteins in protein purification. (a) Glutathione-S-transferase (GST) is a small enzyme that binds glutathione. (b) The GST tag is fused to the carboxyl terminus of the protein by genetic engineering. The tagged protein is expressed in the cell and is present in the crude extract when the cells are lysed. The extract is subjected to affinity chromatography through a matrix with immobilized glutathione.
A shorter tag with widespread application consists of a simple sequence of six or more His residues. These histidine tags, or His tags, bind tightly and specifically to nickel ions. A chromatography matrix with immobilized can be used to quickly separate a His-tagged protein from other proteins in an extract. Some of the larger tags, such as maltose-binding protein, provide added stability and solubility, allowing the purification of cloned proteins that are otherwise inactive due to improper folding or insolubility.
Affinity chromatography using terminal tags is powerful and convenient. The tags have been successfully used in thousands of published studies; in many cases, the protein would be impossible to purify and study without the tag. However, even very small tags can affect the properties of the proteins they are attached to, thereby influencing the study results. For example, the tag may adversely affect protein folding. Even if the tag is removed by a protease, one or a few extra amino acid residues can remain behind on the target protein, which may or may not affect the protein’s activity. The types of experiments to be carried out, and the results obtained from them, should always be evaluated with the aid of well-designed controls to assess any effect of a tag on protein function.
The Polymerase Chain Reaction Offers Many Options for Cloning Experiments
Many adaptations of PCR have increased its utility in cloning. For example, sequences in RNA can be amplified if the first PCR cycle uses reverse transcriptase, an enzyme that works like DNA polymerase (see Fig. 8-33) but uses RNA as a template (Fig. 9-12a). After the DNA strand is made from the RNA template, the remaining cycles can be carried out with DNA polymerases, using standard PCR protocols. This reverse transcriptase PCR (RT-PCR) can be used, for example, to detect sequences derived from living cells (which are transcribing their DNA into RNA) as opposed to dead tissues.
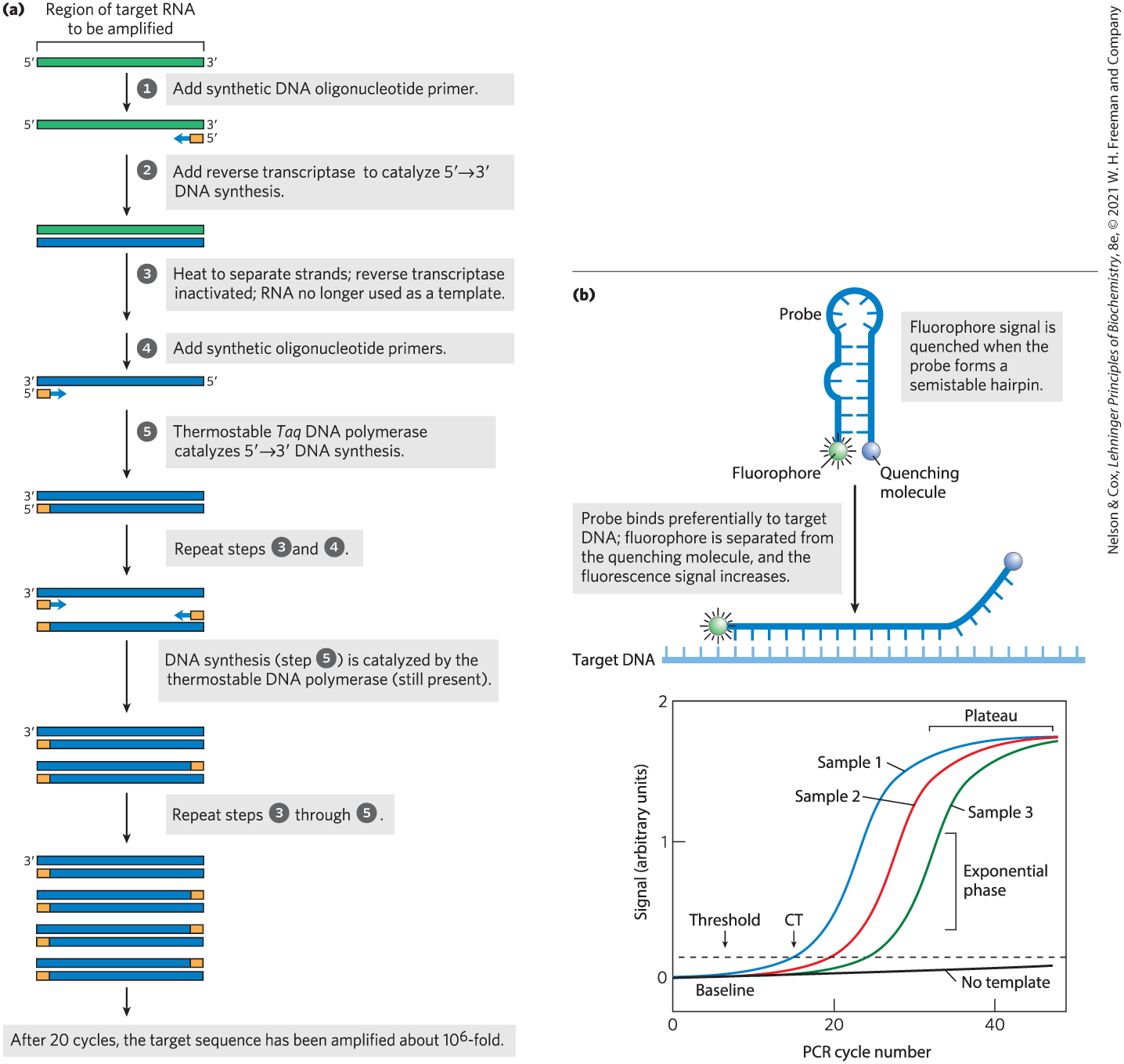
FIGURE 9-12 Some applications of PCR. (a) In reverse transcriptase PCR, or RT-PCR, RNA molecules are amplified by using reverse transcriptase in the first two cycles. (b) In quantitative PCR, or qPCR, careful monitoring of the progress of a PCR amplification allows one to determine when a DNA segment has been amplified to a specified threshold level. The amount of PCR product present is determined by measuring the level of a fluorescent probe attached to a reporter oligonucleotide complementary to the DNA segment that is being amplified. Probe fluorescence is not detectable initially, due to a fluorescence quencher attached to the same oligonucleotide. When the reporter oligonucleotide pairs with its complement in a copy of the amplified DNA segment, the fluorophore is separated from the quenching molecule and fluorescence results. As the PCR reaction proceeds, the amount of the targeted DNA segment increases exponentially, and the fluorescent signal also increases exponentially as the oligonucleotide probes anneal to the amplified segments. After many PCR cycles, the signal reaches a plateau as one or more reaction components become exhausted. When a segment is present in greater amounts in one sample than another, its amplification reaches a defined threshold level earlier. The “No template” line follows the slow increase in background signal observed in a control that does not include added sample DNA. CT is the cycle number at which the threshold is first surpassed.
PCR protocols can also be used to estimate the relative copy numbers of particular sequences in a sample, an approach called quantitative PCR (qPCR) or real-time PCR. If a DNA sequence is present in higher than usual amounts in a sample — for example, if certain genes are amplified in tumor cells — qPCR can reveal the increased representation of that sequence. In brief, the PCR is carried out in the presence of a probe that emits a fluorescent signal when the PCR product is present (Fig. 9-12b). If the sequence of interest is present at higher levels than other sequences in the sample, the PCR signal will reach a predetermined threshold faster. Reverse transcriptase PCR and qPCR can be combined to determine the relative concentrations of a particular mRNA molecule in a cell, and thereby monitor gene expression under different environmental conditions.
DNA Libraries Are Specialized Catalogs of Genetic Information
In some instances, it is useful to clone many genes or genomic segments rather than a particular one. A DNA library is a collection of DNA clones, usually gathered for purposes of gene discovery or the determination of gene or protein function. The library can take a variety of forms, depending on the source of the DNA and the ultimate purpose of the library.
An example is a library that includes only the genes that are transcribed into RNA — expressed — in a given organism or even just in certain cells or tissues. Such a library lacks any genomic DNA that is not transcribed. The researcher first extracts mRNA from an organism, or from specific cells of an organism, and then prepares the complementary DNAs (cDNAs). Like RT-PCR, this multistep reaction (Figure 9-13a) relies on reverse transcriptase, which synthesizes DNA from a template RNA. The resulting double-stranded DNA fragments are inserted into a suitable vector and cloned, creating a population of clones called a cDNA library. If the library host is a bacterium like E. coli, each cell in the population will carry one particular cloned sequence. The library will encompass many millions of cells with millions of different cloned segments. The presence of a gene for a particular protein in such a library implies that this gene is expressed in the cells and under the conditions used to generate the library.
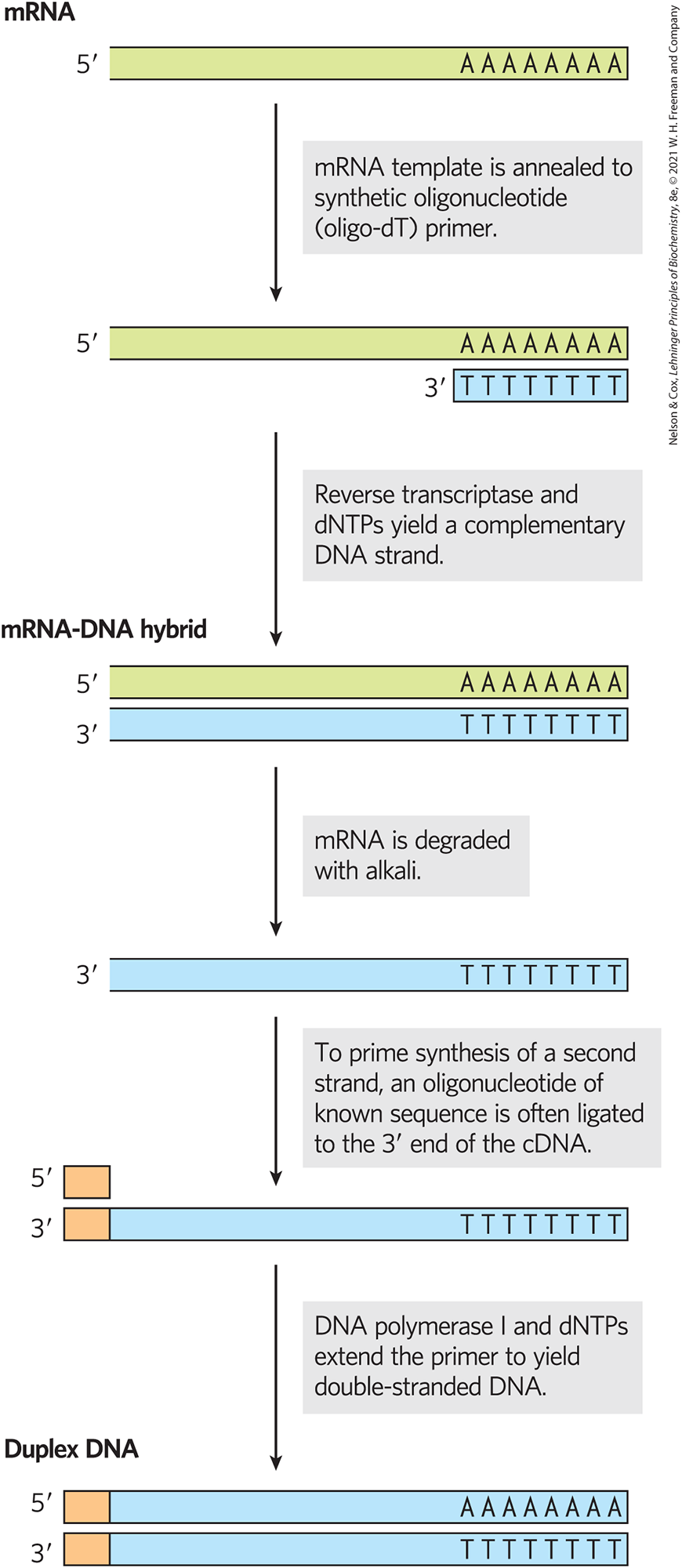
FIGURE 9-13 Building a cDNA library from mRNA. A cell’s total mRNA content includes transcripts from thousands of genes, and the cDNAs generated from this mRNA are correspondingly heterogeneous. Reverse transcriptase can synthesize DNA on an RNA or a DNA template. To prime the synthesis of a second DNA strand, oligonucleotides of known sequence are ligated to the end of the first strand, and the double-stranded cDNA so produced is cloned into a plasmid.
Another type of library, called a combinatorial gene library or simply a gene library, focuses on sequence variants within one gene. For example, beginning with the cloned gene of enzyme X, a segment of the gene could be replaced with nearly identical fragments synthesized with a slight imprecision so that each clone had one or two random base pair changes relative to the original. For example, the gene segment of interest could be amplified by PCR using an altered DNA polymerase that was slightly inaccurate. The library of clones would then consist of many cells, many of which harbored a different variant of the gene for enzyme X. Investigators could use the library to select for variants of enzyme X with enhanced catalytic properties or could simply determine which changes were functional and which were not. The possibilities are limited only by the imagination of the researcher.
SUMMARY 9.1 Studying Genes and Their Products
- DNA cloning and genetic engineering involve the cleavage of DNA and assembly of DNA segments in new combinations — recombinant DNA. Cloning entails generating a DNA fragment of interest, inserting the fragment into a suitable cloning vector, transferring the vector with the DNA insert into a host cell for replication, and identifying and selecting cells that contain the DNA fragment.
- Key enzymes in gene cloning include restriction endonucleases (especially the type II enzymes) and DNA ligase.
- Cloning vectors include plasmids and, for the longest DNA inserts, bacterial artificial chromosomes (BACs) and yeast artificial chromosomes (YACs).
- Cloned genes can be expressed in a host cell by incorporating them into expression vectors that have the sequence signals needed for transcription and translation.
- Proteins can be expressed in different types of cells using expression systems with various useful features and advantages.
- Genetic engineering techniques can alter cloned genes as required by the investigator.
- Proteins or peptides can be attached to a protein of interest by altering its cloned gene, creating a fusion protein. The additional peptide segments can be used to detect the protein or to purify it, using convenient affinity chromatography methods.
- The polymerase chain reaction (PCR) permits the amplification of chosen segments of DNA or RNA for cloning and can be adapted to determine gene copy number or to monitor gene expression quantitatively.
- DNA libraries consist of many clones, encompassing many genomic segments or many variants of a particular gene.
 DNA cloning and genetic engineering involve the cleavage of DNA and assembly of DNA segments in new combinations — recombinant DNA. Cloning entails generating a DNA fragment of interest, inserting the fragment into a suitable cloning vector, transferring the vector with the DNA insert into a host cell for replication, and identifying and selecting cells that contain the DNA fragment.
DNA cloning and genetic engineering involve the cleavage of DNA and assembly of DNA segments in new combinations — recombinant DNA. Cloning entails generating a DNA fragment of interest, inserting the fragment into a suitable cloning vector, transferring the vector with the DNA insert into a host cell for replication, and identifying and selecting cells that contain the DNA fragment.