25.1 DNA Replication
Long before the structure of DNA was known, scientists wondered at the ability of organisms to create faithful copies of themselves and, later, at the ability of cells to produce many identical copies of large, complex macromolecules. Speculation about these problems centered around the concept of a template, a structure that would allow molecules to be lined up in a specific order and joined to create a macromolecule with a unique sequence and function. The 1940s brought the revelation that DNA was the genetic molecule, but not until James Watson and Francis Crick deduced its structure did the way in which DNA could act as a template for the replication and transmission of genetic information become clear: one strand is the complement of the other. The strict base-pairing rules mean that each strand provides the template for a new strand with a predictable and complementary sequence (see Figs. 8-14, 8-15). The fundamental properties of the DNA replication process and the mechanisms used by the enzymes that catalyze it have proved to be essentially identical in all species.
 Long before the structure of DNA was known, scientists wondered at the ability of organisms to create faithful copies of themselves and, later, at the ability of cells to produce many identical copies of large, complex macromolecules. Speculation about these problems centered around the concept of a
Long before the structure of DNA was known, scientists wondered at the ability of organisms to create faithful copies of themselves and, later, at the ability of cells to produce many identical copies of large, complex macromolecules. Speculation about these problems centered around the concept of a DNA Replication Follows a Set of Fundamental Rules
Early research on bacterial DNA replication and its enzymes helped to establish several basic properties that have proven applicable to DNA synthesis in every organism.
DNA Replication Is Semiconservative
Each DNA strand serves as a template for the synthesis of a new strand, producing two new DNA molecules, each with one new strand and one old strand. This is semiconservative replication. The semiconservative nature of replication was established by Matthew Meselson and Frank Stahl in 1957.
Replication Begins at an Origin and Usually Proceeds Bidirectionally
Following the confirmation of a semiconservative mechanism of replication, a host of questions arose. Are the parent DNA strands completely unwound before each is replicated? Does replication begin at random places or at a unique point? After initiation at any point in the DNA, does replication proceed in one direction or both?
Photographic images of tritium –labeled bacterial DNA made by John Cairns revealed that the intact chromosome of E. coli is a single huge circle, 1.7 mm long. Radioactive DNA isolated from cells during replication showed an extra loop (Fig. 25-1). Cairns concluded that the loop resulted from the formation of two radioactive daughter strands, each complementary to a parent strand. One or both ends of the loop are dynamic points, termed replication forks, where parent DNA is being unwound and the separated strands quickly replicated. Cairns’s results demonstrated that both DNA strands are replicated simultaneously, and variations on his experiment showed that replication of bacterial chromosomes is bidirectional: both ends of the loop have active replication forks.
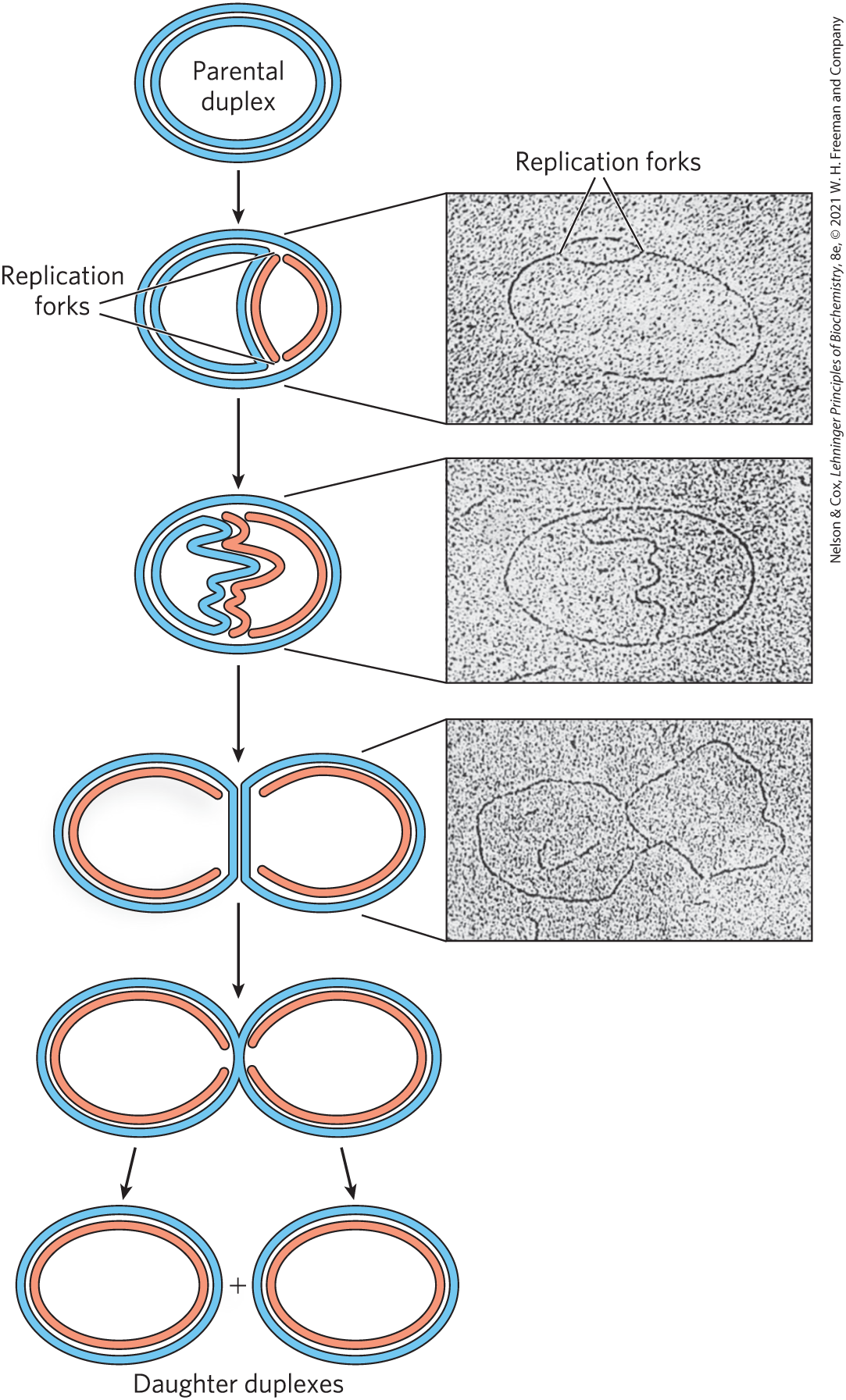
FIGURE 25-1 Visualization of DNA replication. Stages in the replication of circular DNA molecules have been visualized by electron microscopy. Replication of a circular chromosome produces a structure resembling the Greek letter theta, , as both strands are replicated simultaneously (new strands shown in light red). The electron micrographs show images of plasmid DNA being replicated from a single replication origin. [Electron micrographs: Cairns, J. (1963), The Chromosome of Escherichia coli, Cold Spring Harbor Symp Quant Biol., 28, 43–46. © Cold Spring Harbor Laboratory Press.]
Determination of whether the replication loops originate at a unique point in the DNA required landmarks along the DNA molecule. These landmarks were provided by a technique called denaturation mapping, developed by Ross Inman and colleagues. Using the 48,502 bp chromosome of bacteriophage , Inman showed that DNA could be selectively denatured at sequences unusually rich in base pairs, generating a reproducible pattern of single-strand bubbles (see Fig. 8-28). Using the denatured regions as points of reference, investigators have subsequently been able to measure the position and progress of the replication forks. Inman and colleagues found that the replication loops always initiated at a unique point, which was termed an origin. They also confirmed the earlier observation that replication is usually bidirectional. For circular DNA molecules, the two replication forks meet at a point on the side of the circle opposite to the origin. Specific origins of replication have since been identified and characterized in bacteria and eukaryotes.
DNA Synthesis Proceeds in a Direction and Is Semidiscontinuous
A new strand of DNA is always synthesized in the direction, with the free as the point at which the DNA is elongated. (Recall from Chapter 8 that the end lacks a nucleotide attached to the position, and the end lacks a nucleotide attached to the position.) Because the two DNA strands are antiparallel, the strand serving as the template is read from its end toward its end.
If synthesis always proceeds in the direction, how can both strands be synthesized simultaneously? If both strands were synthesized continuously while the replication fork moved, one strand would have to undergo synthesis. This problem was resolved by Reiji Okazaki and colleagues in the 1960s. Okazaki found that one of the new DNA strands is synthesized in short pieces, now called Okazaki fragments. Thus, one strand is synthesized continuously and the other discontinuously (Fig. 25-2). The continuous strand, or leading strand, is the one for which synthesis proceeds in the same direction that the replication fork moves. The discontinuous strand, or lagging strand, is the one in which synthesis proceeds in the direction opposite to the direction of fork movement. Okazaki fragments are typically 150 to 200 nucleotides long in eukaryotes, and 1,000 to 2,000 nucleotides long in bacteria. As we shall see, leading and lagging strand syntheses are tightly coordinated.
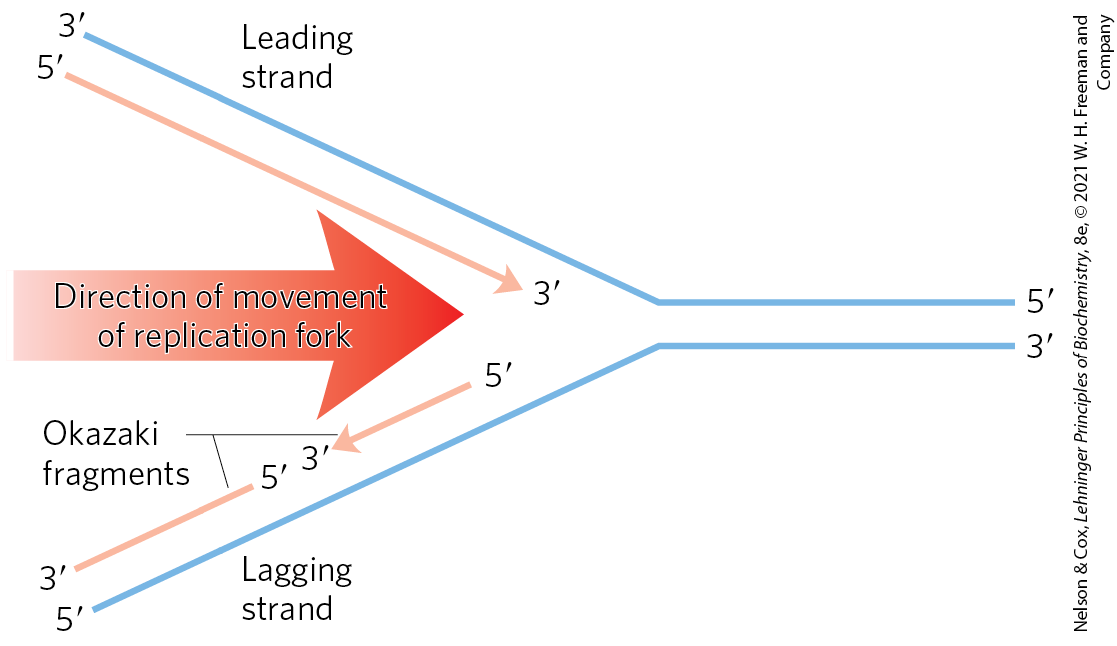
FIGURE 25-2 Defining DNA strands at the replication fork. A new DNA strand (light red) is always synthesized in the direction. The template is read in the opposite direction, . The leading strand is continuously synthesized in the direction taken by the replication fork. The other strand, the lagging strand, is synthesized discontinuously in short pieces (Okazaki fragments) in a direction opposite to that in which the replication fork moves. The Okazaki fragments are spliced together by DNA ligase. In bacteria, Okazaki fragments are ∼1,000 to 2,000 nucleotides long. In eukaryotic cells, they are 150 to 200 nucleotides long.
DNA Is Degraded by Nucleases
To explain the enzymology of DNA replication, we first introduce the enzymes that degrade DNA rather than synthesize it. These enzymes are known as nucleases, or DNases if they are specific for DNA rather than RNA. Every cell contains several different nucleases, belonging to two broad classes: exonucleases and endonucleases. Exonucleases degrade nucleic acids from one end of the molecule. Many operate in only the direction or the direction, removing nucleotides only from the end or the end, respectively, of one strand of a double-stranded nucleic acid or of a single-stranded DNA. Endonucleases can begin to degrade at specific internal sites in a nucleic acid strand or molecule, reducing it to smaller and smaller fragments. A few exonucleases and endonucleases degrade only single-stranded DNA. A few important classes of endonucleases cleave only at specific nucleotide sequences (such as the restriction endonucleases that are so important in biotechnology; see Chapter 9, Fig. 9-2). You will encounter many types of nucleases in this and subsequent chapters.
DNA Is Synthesized by DNA Polymerases

Arthur Kornberg, 1918–2007
The search for an enzyme that could synthesize DNA began in 1955. Work by Arthur Kornberg and colleagues led to the purification and characterization of a DNA polymerase from E. coli cells, a single-polypeptide enzyme now called DNA polymerase I (; encoded by the polA gene). Much later, investigators found that E. coli contains at least four additional distinct DNA polymerases, described below.
Detailed studies of DNA polymerase I revealed features of the DNA synthetic process that are now known to be common to all DNA polymerases. The fundamental reaction is a phosphoryl group transfer. The nucleophile is the -hydroxyl group of the nucleotide at the end of the growing strand. Nucleophilic attack occurs at the α phosphorus of the incoming deoxynucleoside -triphosphate (Fig. 25-3). Inorganic pyrophosphate is released in the reaction. The general reaction is
(25-1)
where dNMP and dNTP are a deoxynucleoside -monophosphate and -triphosphate, respectively. Catalysis by virtually all DNA polymerases prominently involves two ions at the active site. One of these helps to deprotonate the -hydroxyl group, rendering it a more effective nucleophile. The other binds to the incoming dNTP and facilitates departure of the pyrophosphate.
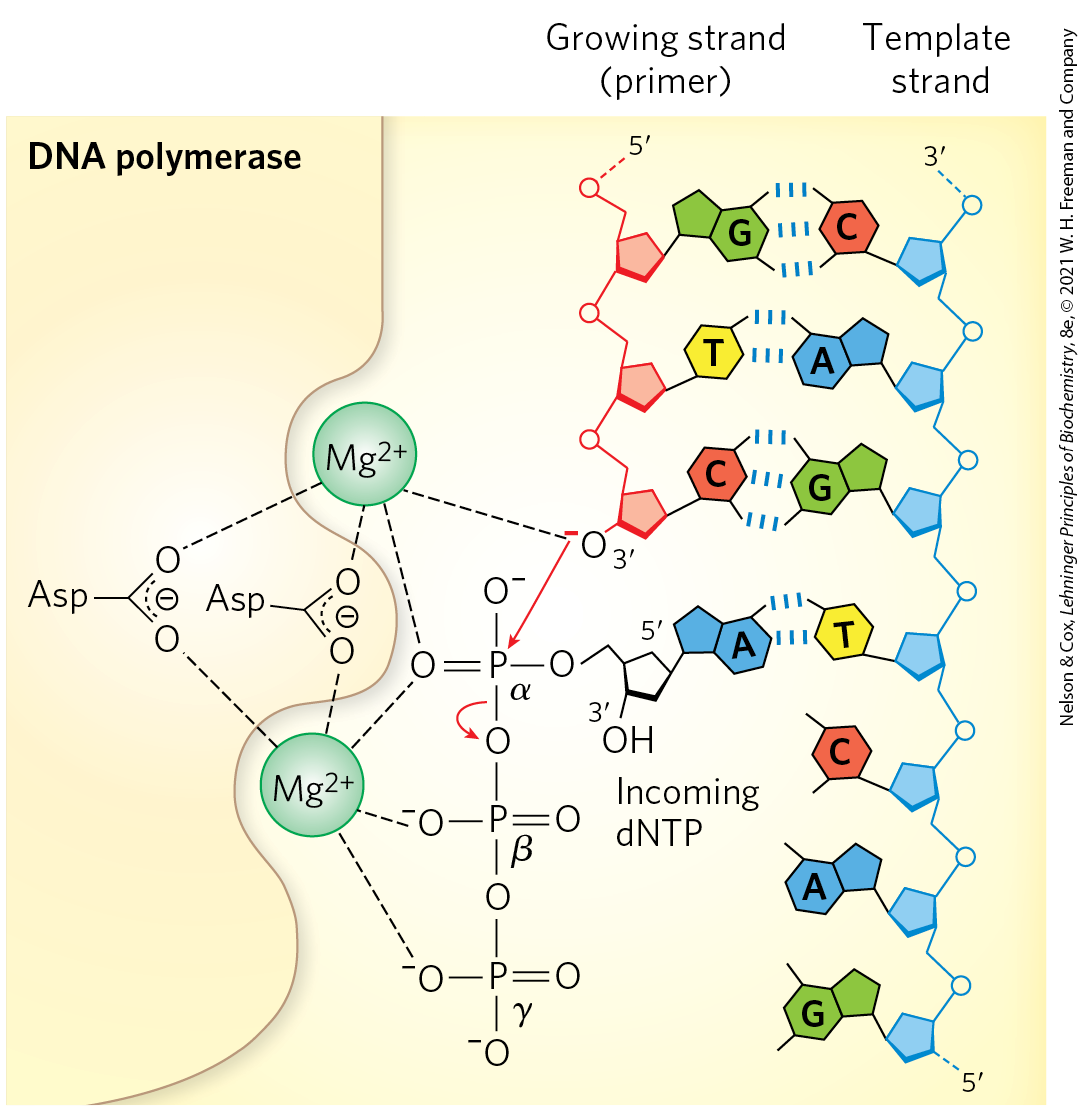
MECHANISM FIGURE 25-3 The DNA polymerase reaction. The catalytic mechanism for addition of a new nucleotide by DNA polymerase involves two ions, coordinated to the phosphate groups of the incoming nucleotide triphosphate, the -hydroxyl group that will act as a nucleophile, and three Asp residues, two of which are highly conserved in all DNA polymerases. The ion depicted at the top facilitates attack of the -hydroxyl group of the primer on the α phosphate of the nucleotide triphosphate; the other ion facilitates displacement of the pyrophosphate. Both ions stabilize the structure of the pentacovalent transition state. RNA polymerases use a similar mechanism.
The reaction seems to proceed with only a minimal change in free energy, given that one phosphodiester bond is formed at the expense of a somewhat less stable phosphate anhydride. However, noncovalent base-stacking and base-pairing interactions provide additional stabilization to the lengthened DNA product relative to the free nucleotide. Also, the formation of products is facilitated in the cell by the 19 kJ/mol generated in the subsequent hydrolysis of the pyrophosphate product by the enzyme pyrophosphatase (p. 485).
 The reaction seems to proceed with only a minimal change in free energy, given that one phosphodiester bond is formed at the expense of a somewhat less stable phosphate anhydride. However, noncovalent base-stacking and base-pairing interactions provide additional stabilization to the lengthened DNA product relative to the free nucleotide. Also, the formation of products is facilitated in the cell by the 19 kJ/mol generated in the subsequent hydrolysis of the pyrophosphate product by the enzyme pyrophosphatase (
The reaction seems to proceed with only a minimal change in free energy, given that one phosphodiester bond is formed at the expense of a somewhat less stable phosphate anhydride. However, noncovalent base-stacking and base-pairing interactions provide additional stabilization to the lengthened DNA product relative to the free nucleotide. Also, the formation of products is facilitated in the cell by the 19 kJ/mol generated in the subsequent hydrolysis of the pyrophosphate product by the enzyme pyrophosphatase (Early work on DNA polymerase I led to the definition of two central requirements for DNA polymerization (Fig. 25-4). First, all DNA polymerases require a template. The polymerization reaction is guided by a template DNA strand according to the base-pairing rules predicted by Watson and Crick: where a guanine is present in the template, a cytosine deoxynucleotide is added to the new strand, and so on. This was a particularly important discovery, not only because it provided a chemical basis for accurate semiconservative DNA replication, but also because it represented the first example of the use of a template to guide a biosynthetic reaction.
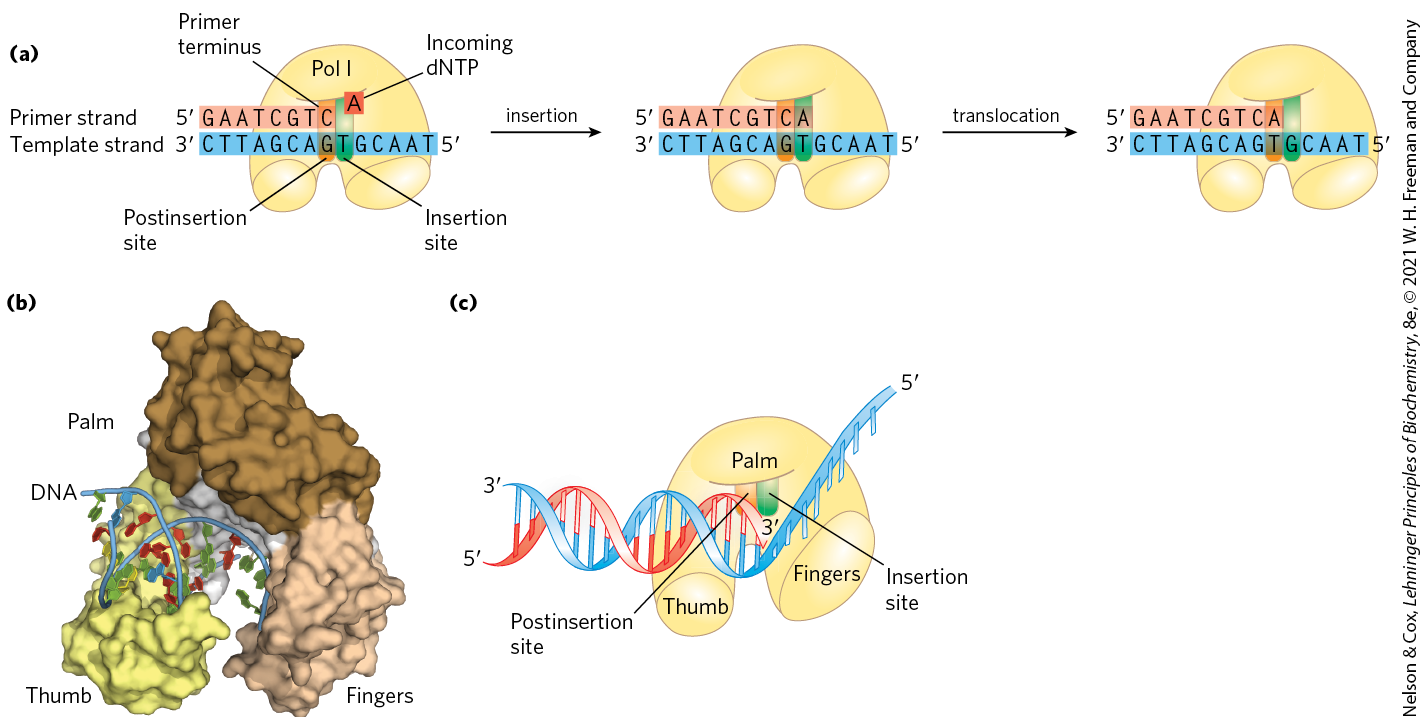
FIGURE 25-4 Elongation of a DNA chain. (a) DNA polymerase I activity requires a single unpaired strand to act as template and a primer strand to provide the free hydroxyl group at the end to which the new nucleotide unit is added. Each incoming complementary nucleotide is bound selectively, in part by base-pairing to the appropriate nucleotide in the template strand. The reaction product has a new free hydroxyl, allowing the addition of another nucleotide. The newly formed base pair translocates to make the active site available to the next pair to be formed. (b) The core of most DNA polymerases is shaped like a human hand that wraps around the active site. The structure shown here is DNA polymerase I of Thermus aquaticus, bound to DNA. (c) A cartoon interpretation shows the insertion site, where the nucleotide addition occurs, and the postinsertion site, to which the newly formed base pair translocates. [(b) Data from PDB ID 4KTQ, Y. Li et al., EMBO J. 17:7514, 1998.]
Second, the polymerases require a primer. A primer is a strand segment (complementary to the template) with a free -hydroxyl group to which a nucleotide can be added; the free end of the primer is called the primer terminus (Fig. 25-4a). In other words, part of the new strand must already be in place: all DNA polymerases can add nucleotides only to a preexisting strand. Many primers are oligonucleotides of RNA rather than DNA, and specialized enzymes synthesize primers when and where they are required.
A DNA polymerase active site has two parts (Fig. 25-4a). The incoming nucleotide is initially positioned in the insertion site. Once the phosphodiester bond is formed, the polymerase slides forward on the DNA and the new base pair is positioned in the postinsertion site. These sites are located in a pocket that resembles the palm of a hand (Fig. 25-4b, c).
After adding a nucleotide to a growing DNA strand, a DNA polymerase either dissociates or moves along the template and adds another nucleotide. Dissociation and reassociation of the polymerase can limit the overall polymerization rate — the process is generally faster when a polymerase adds more nucleotides without dissociating from the template. The average number of nucleotides added before a polymerase dissociates defines its processivity. DNA polymerases vary greatly in processivity; some add just a few nucleotides before dissociating, whereas others add many thousands.
Replication Is Very Accurate
Replication proceeds with an extraordinary degree of fidelity. In E. coli, a mistake is made only once for every to nucleotides added. For the E. coli chromosome of bp, this means that an error occurs only once per 1,000 to 10,000 replications. During polymerization, discrimination between correct and incorrect nucleotides relies not just on the hydrogen bonds that specify the correct pairing between complementary bases but also on the common geometry of the standard and base pairs (Fig. 25-5). The active site of DNA polymerase I accommodates only base pairs with this geometry. An incorrect nucleotide may be able to hydrogen-bond with a base in the template, but it generally will not fit into the active site. Incorrect bases can be rejected before the phosphodiester bond is formed.
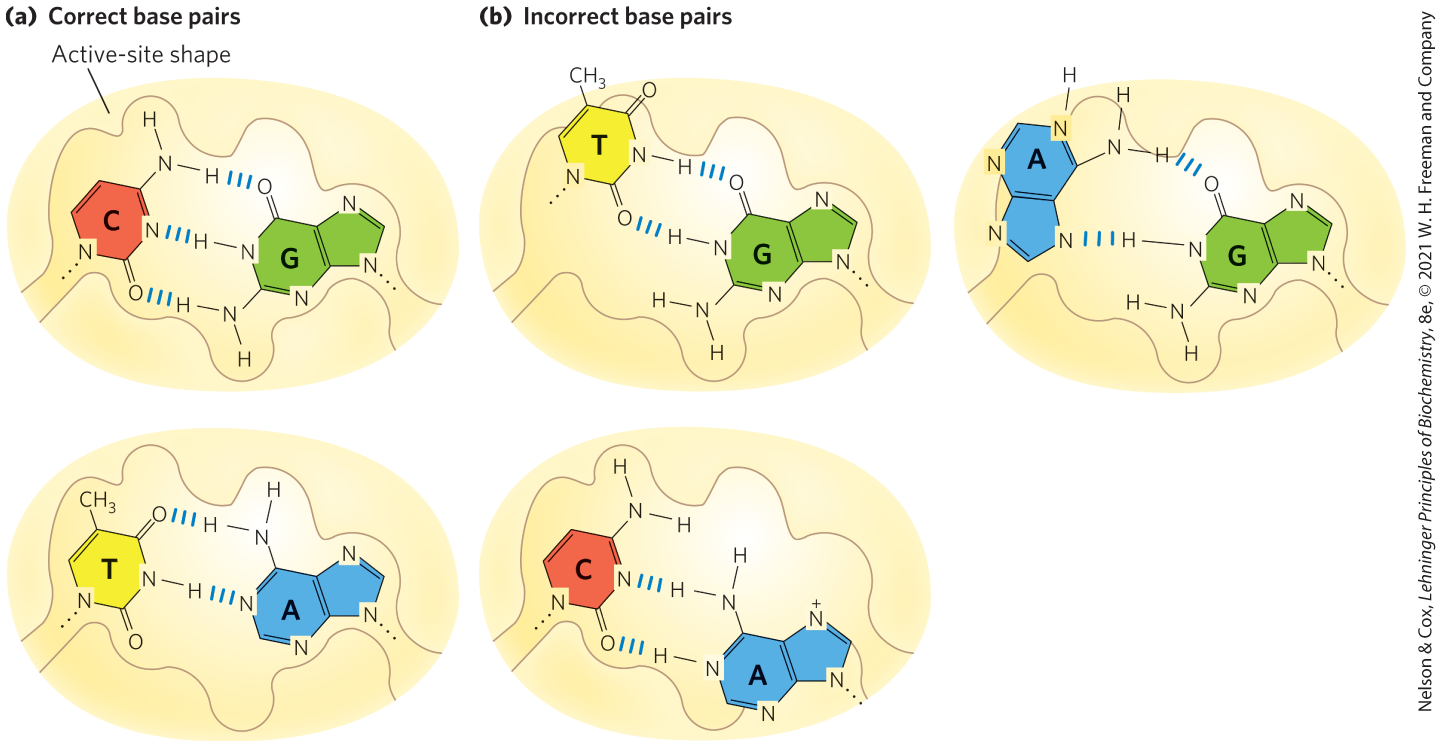
FIGURE 25-5 Contribution of base-pair geometry to the fidelity of DNA replication. (a) The standard and base pairs have very similar geometries, and an active site sized to fit one will generally accommodate the other. (b) The geometry of incorrectly paired bases can exclude them from the active site, as occurs on DNA polymerase.
The accuracy of the polymerization reaction itself, however, is insufficient to account for the high degree of fidelity in replication. Careful measurements in vitro have shown that DNA polymerases insert one incorrect nucleotide for every correct ones. These mistakes sometimes occur because a base is briefly in an unusual tautomeric form (see Fig. 8-9), allowing it to hydrogen-bond with an incorrect partner. In vivo, the error rate is reduced by additional enzymatic mechanisms.
One mechanism intrinsic to many DNA polymerases is a separate exonuclease activity that double-checks each nucleotide after it is added. This nuclease activity permits the enzyme to remove a newly added nucleotide and is highly specific for mismatched base pairs (Fig. 25-6). If the polymerase has added the wrong nucleotide, translocation of the enzyme to the position where the next nucleotide is to be added is inhibited. This kinetic pause provides the opportunity for a correction. The exonuclease activity cleaves the most recently added phosphodiester bond and removes the mispaired nucleotide; the polymerase then adds another nucleotide to begin active synthesis again. This activity, known as proofreading, is not simply the reverse of the polymerization reaction (Eqn 25-1). Instead, replacement of the incorrect nucleotide requires the expenditure of three high-energy bonds. The polymerizing and proofreading activities of a DNA polymerase can be measured separately. Proofreading improves the inherent accuracy of the polymerization reaction . In the monomeric DNA polymerase I, the polymerizing and proofreading activities have separate active sites within the same polypeptide.
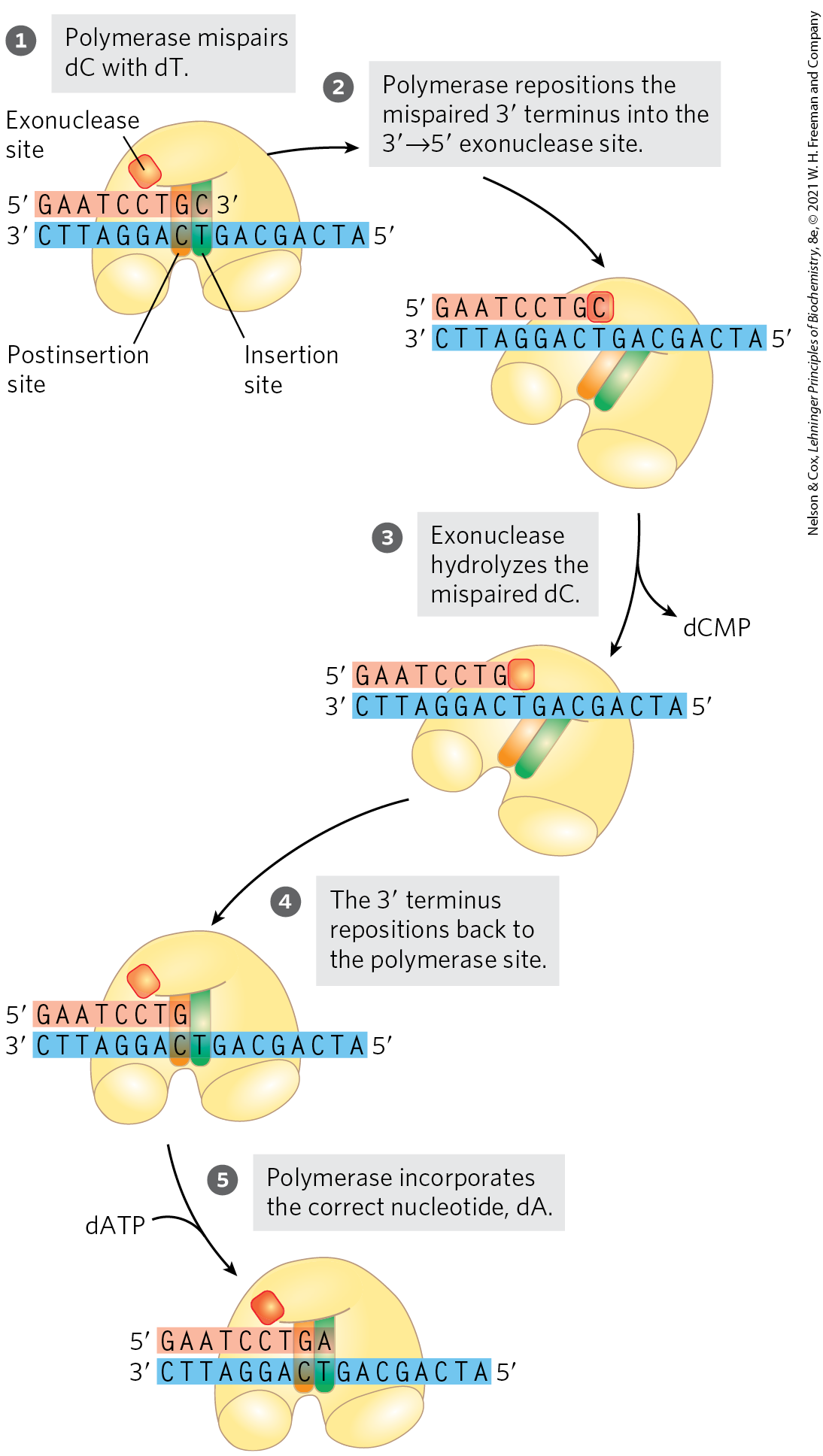
FIGURE 25-6 An example of error correction by the exonuclease activity of DNA polymerase I. Structural analysis has located the exonuclease activity behind the polymerase activity as the enzyme is oriented in its movement along the DNA. A mismatched base (here, a C–T mismatch) impedes translocation of DNA polymerase I (Pol I) to the next site.
When base selection and proofreading are combined, DNA polymerase leaves behind one net error for every bases added. Yet the measured accuracy of replication in E. coli is higher still. The additional accuracy is provided by a separate enzyme system that repairs the mismatched base pairs remaining after replication. We describe this mismatch repair, along with other DNA repair processes, in Section 25.2.
 When base selection and proofreading are combined, DNA polymerase leaves behind one net error for every bases added. Yet the measured accuracy of replication in E. coli is higher still. The additional accuracy is provided by a separate enzyme system that repairs the mismatched base pairs remaining after replication. We describe this mismatch repair, along with other DNA repair processes, in
When base selection and proofreading are combined, DNA polymerase leaves behind one net error for every bases added. Yet the measured accuracy of replication in E. coli is higher still. The additional accuracy is provided by a separate enzyme system that repairs the mismatched base pairs remaining after replication. We describe this mismatch repair, along with other DNA repair processes, in E. coli Has at Least Five DNA Polymerases
More than 90% of the DNA polymerase activity observed in E. coli extracts can be accounted for by DNA polymerase I. Soon after the isolation of this enzyme in 1955, however, evidence began to accumulate that it is not suited for replication of the large E. coli chromosome. First, the rate at which it adds nucleotides (600 nucleotides/min) is too slow (by a factor of 100 or more) to account for the rates at which the replication fork moves in the bacterial cell. Second, DNA polymerase I has a relatively low processivity. Third, genetic studies have demonstrated that many genes, and therefore many proteins, are involved in replication: DNA polymerase I clearly does not act alone. Fourth, and most important, in 1969 John Cairns isolated a bacterial strain with an altered gene for DNA polymerase I that produced an inactive enzyme. Although this strain was abnormally sensitive to agents that damaged DNA, it was nevertheless viable.
A search for other DNA polymerases led to the discovery of E. coli DNA polymerase II and DNA polymerase III in the early 1970s. DNA polymerase II is an enzyme involved in one type of DNA repair (Section 25.3). DNA polymerase III is the principal replication enzyme in E. coli. DNA polymerases IV and V, identified in 1999, are involved in an unusual form of DNA repair (Section 25.2). The properties of these five DNA polymerases are compared in Table 25-1.
| DNA polymerase | ||||||
| I | IIa | III | IVa | Va | ||
|---|---|---|---|---|---|---|
Structural geneb |
polA |
polB |
polC (dnaE) |
dinB |
umuC |
|
Subunits (number of different types) |
1 |
7 |
9 |
1 |
3 |
|
103,000 |
88,000c |
1,065,400 |
39,100 |
110,000 |
||
exonuclease (proofreading) |
Yes |
Yes |
Yes |
No |
No |
|
exonuclease |
Yes |
No |
No |
No |
No |
|
Polymerization rate (nucleotides/s) |
10–20 |
40 |
250–1,000 |
2–3 |
1 |
|
Processivity (nucleotides added before polymerase dissociates) |
3–200 |
1,500 |
≥500,000 |
1 |
6–8 |
|
|
||||||
DNA polymerase I, then, is not the primary enzyme of replication; instead, it performs a host of cleanup functions during replication, recombination, and repair. The polymerase’s special functions are enhanced by its exonuclease activity. This activity, distinct from the proofreading exonuclease (Fig. 25-6), is located in a structural domain that can be separated from the rest of the enzyme by mild protease treatment. When the exonuclease domain is removed, the remaining fragment , the large fragment or Klenow fragment, retains the polymerization and proofreading activities. The exonuclease activity of intact DNA polymerase I can replace a segment of DNA (or RNA) paired to the template strand, in a process known as nick translation (Fig. 25-7). Most other DNA polymerases lack a exonuclease activity.
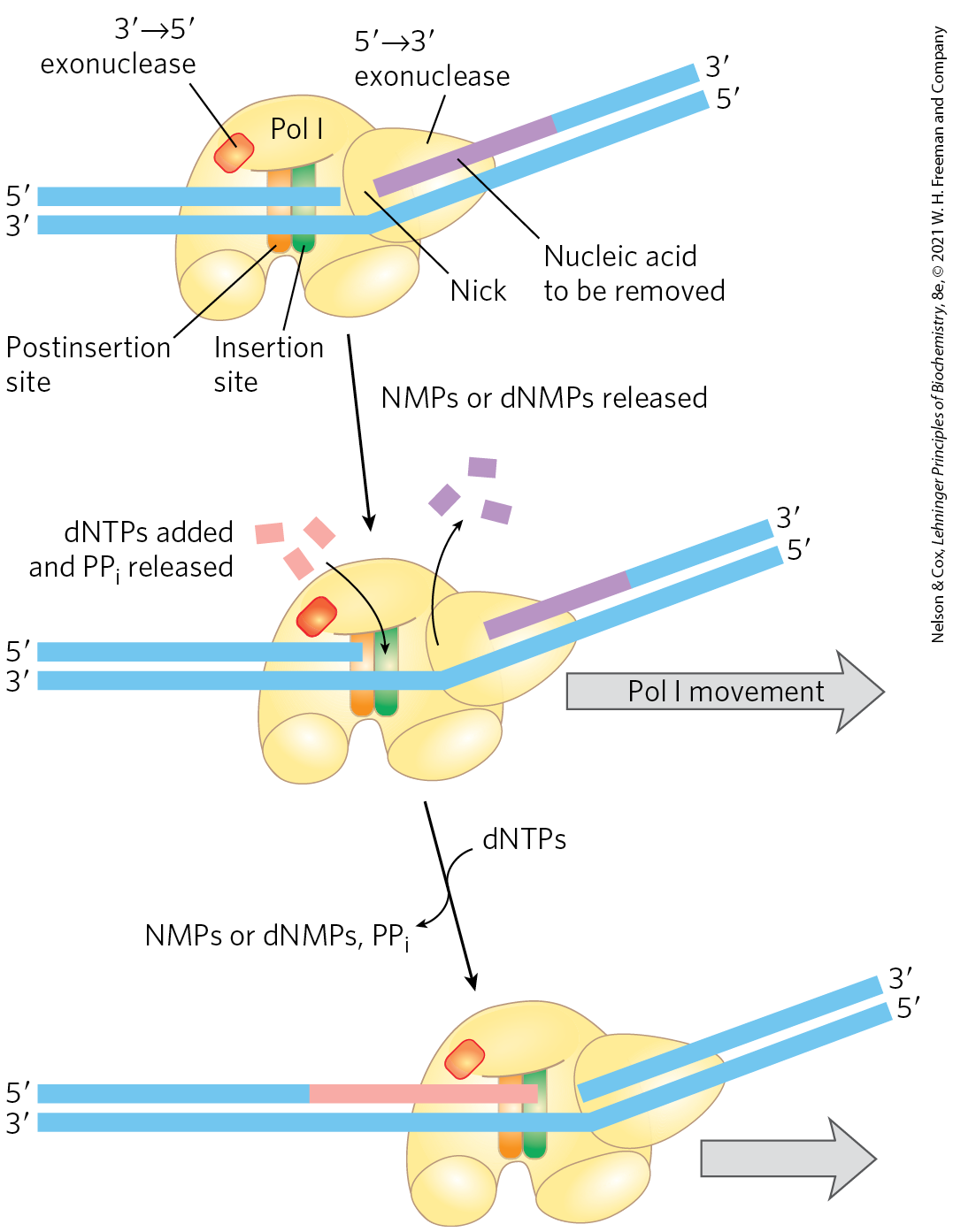
FIGURE 25-7 Nick translation. The bacterial DNA polymerase I has three domains, catalyzing its DNA polymerase, exonuclease, and exonuclease activities. The exonuclease domain is in front of the enzyme as it moves along the DNA and is not shown in Figure 25-4. By degrading the DNA strand ahead of the enzyme and synthesizing a new strand behind, DNA polymerase I can promote nick translation, in which a break or nick in the DNA is effectively moved along with the enzyme. This process has a role in DNA repair and in the removal of RNA primers during replication (both described later in this chapter). The strand of nucleic acid to be removed (either DNA or RNA) is shown in purple, the replacement strand in red. DNA synthesis begins at a nick (a broken phosphodiester bond, leaving a free hydroxyl and a free phosphate). A nick remains where DNA polymerase I eventually dissociates, and the nick is later sealed by another enzyme.
DNA polymerase III is much more complex than DNA polymerase I, with nine different kinds of subunits (Table 25-2). Its polymerization and proofreading activities reside in its α and ε subunits, respectively. The subunit associates with α and ε to form a core polymerase, which can polymerize DNA but with limited processivity. Up to three core polymerases can be linked by a clamp-loading complex consisting of five subunits of three different types, . The core polymerases are linked through the (tau) subunits. Two additional subunits, (chi) and (psi), are bound to the clamp-loading complex. The entire assembly of 16 protein subunits (eight different types) is called DNA polymerase III* (Fig. 25-8a).
 |
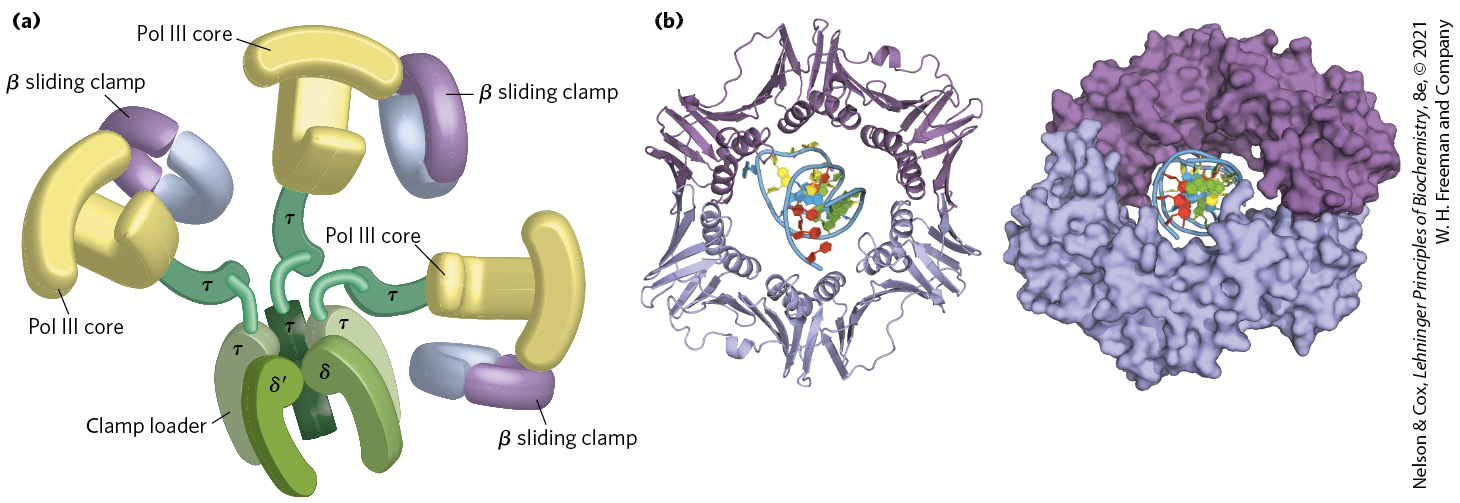
FIGURE 25-8 DNA polymerase III. (a) Architecture of bacterial DNA polymerase III (Pol III). Three core domains, composed of subunits α, ε, and , are linked by a five-subunit clamp-loading complex, with the composition . The core subunits and clamp-loading complex constitute DNA polymerase III*. The other two subunits of DNA polymerase III*, and (not shown), also bind to the clamp-loading complex. Three β clamps interact with the three core subassemblies, each clamp a dimer of the β subunit. The complex interacts with the DnaB helicase (described later in the text) through the subunits. (b) Two β subunits of E. coli polymerase III form a circular clamp that surrounds the DNA. The clamp slides along the DNA molecule, increasing the processivity of the polymerase III holoenzyme to more than 500,000 nucleotides by preventing its dissociation from the DNA. The two β subunits are shown in two shades of purple as ribbon structures (left) and surface contour images (right), surrounding the DNA. [(a) Information from N. Yao and M. O’Donnell, Mol. Biosyst. 4:1075, 2008. (b) Data from PDB ID 2POL, X.-P. Kong et al., Cell 69:425, 1992.]
DNA polymerase III* can polymerize DNA, but with a much lower processivity than one would expect for the organized replication of an entire chromosome. The necessary increase in processivity is provided by the addition of the β subunits. The β subunits associate in pairs to form donut-shaped structures that encircle the DNA and act like clamps (Fig. 25-8b). Each dimer associates with a core subassembly of polymerase III* (one dimeric clamp per active core subassembly) and slides along the DNA as replication proceeds. The β sliding clamp prevents the dissociation of DNA polymerase III from DNA, dramatically increasing processivity — to greater than 500,000 (Table 25-1). The addition of the β subunits converts DNA polymerase III* to DNA polymerase III holoenzyme.
DNA Replication Requires Many Enzymes and Protein Factors
Replication in E. coli requires not just a single DNA polymerase but 20 or more different enzymes and proteins, each performing a specific task. The entire complex has been termed the DNA replicase system or replisome. The enzymatic complexity of replication reflects the constraints imposed by the structure of DNA and by the requirements for accuracy. The main classes of replication enzymes are considered here in terms of the problems they overcome.
The DNA must be separated into two strands that each act as a template. This is generally accomplished by helicases, enzymes that move along the DNA and separate the strands, using chemical energy from ATP. Strand separation creates topological stress in the helical DNA structure (see Fig. 24-11), which is relieved by the action of topoisomerases. The separated strands are stabilized by DNA-binding proteins. As noted earlier, before DNA polymerases can begin synthesizing DNA, primers must be present on the template — generally, short segments of RNA synthesized by enzymes known as primases. Ultimately, the RNA primers are removed and replaced by DNA; in E. coli, this is one of the many functions of DNA polymerase I. A specialized nuclease that degrades RNA in RNA-DNA hybrids, called RNase H1, also removes some RNA primers. After an RNA primer is removed and the gap is filled in with DNA, a nick remains in the DNA backbone in the form of a broken phosphodiester bond. These nicks are sealed by DNA ligases. All these processes require coordination and regulation best characterized in the E. coli system.
Replication of the E. coli Chromosome Proceeds in Stages
The synthesis of a DNA molecule can be divided into three stages: initiation, elongation, and termination, distinguished both by the reactions taking place and by the enzymes required. As you will find here and in the next two chapters, synthesis of the major information-containing biological polymers — DNAs, RNAs, and proteins — can be understood in terms of these same three stages, with the stages of each pathway having unique characteristics. The events described below reflect information derived primarily from in vitro experiments using purified E. coli proteins, although the principles are highly conserved in all replication systems.
Initiation
The E. coli replication origin, oriC, consists of 245 bp and contains DNA sequence elements that are highly conserved among bacterial replication origins. The general arrangement of the conserved sequences is illustrated in Figure 25-9. Two types of sequences are of special interest: five repeats of a 9 bp sequence (R sites) that serve as binding sites for the key initiator protein, DnaA, and a region rich in base pairs called the DNA unwinding element (DUE). There are three additional DnaA-binding sites (I sites), and binding sites for the proteins IHF (integration host factor) and FIS (factor for inversion stimulation). These two proteins were discovered as necessary components of certain recombination reactions described later in this chapter, and their names reflect those roles. Another DNA-binding protein, HU (a histonelike bacterial protein originally dubbed factor U), also participates but does not have a specific binding site.
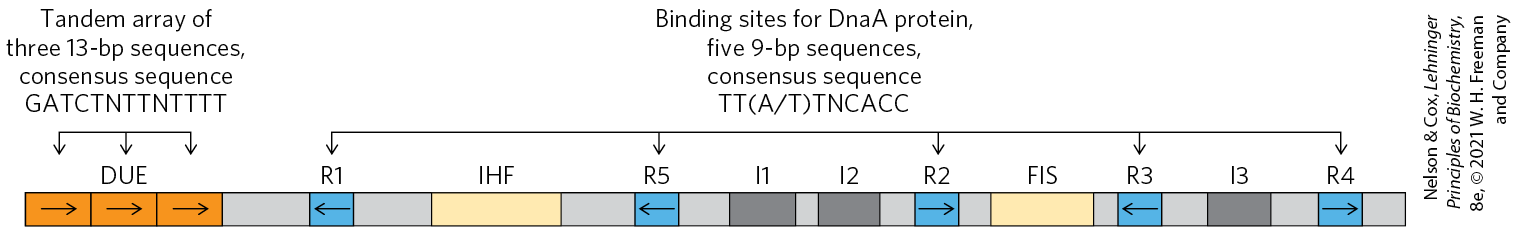
FIGURE 25-9 Arrangement of sequences in the E. coli replication origin, oriC. Conserved sequences for key repeated elements are shown. N represents any of the four nucleotides. The horizontal arrows indicate the orientations of the nucleotide sequences (left-to-right arrow denotes a sequence in the top strand; right-to-left, in the bottom strand). FIS and IHF are binding sites for proteins described in the text. R sites are bound by DnaA. I sites are additional DnaA-binding sites (with different sequences, labeled I1, I2, and I3), for DnaA only when the protein is complexed with ATP.
At least 10 different enzymes or proteins (summarized in Table 25-3) participate in the initiation phase of replication. They open the DNA helix at the origin and establish a prepriming complex for subsequent reactions. The crucial component in the initiation process is the DnaA protein, a member of the protein family (ATPases associated with diverse cellular activities). Many , including DnaA, form oligomers and hydrolyze ATP relatively slowly. This ATP hydrolysis acts as a switch that mediates interconversion of the protein between two states. In the case of DnaA, the ATP-bound form is active and the ADP-bound form is inactive.
| Protein | Number of subunits | Function | |
|---|---|---|---|
DnaA protein |
52,000 |
1 |
Recognizes oriC sequence; opens duplex at specific sites in origin |
DnaB protein (helicase) |
300,000 |
6a |
Unwinds DNA |
DnaC protein |
174,000 |
6a |
Required for DnaB binding at origin |
HU |
19,000 |
2 |
Histonelike protein; DNA-binding protein; stimulates initiation |
FIS |
22,500 |
2a |
DNA-binding protein; stimulates initiation |
IHF |
22,000 |
2 |
DNA-binding protein; stimulates initiation |
Primase (DnaG protein) |
60,000 |
1 |
Synthesizes RNA primers |
Single-stranded DNA–binding protein (SSB) |
75,600 |
4a |
Binds single-stranded DNA |
DNA gyrase (DNA topoisomerase II) |
400,000 |
4 |
Relieves torsional strain generated by DNA unwinding |
Dam methylase |
32,000 |
1 |
Methylates GATC sequences at oriC |
aSubunits in these cases are identical. |
|||
Eight DnaA protein molecules, all in the ATP-bound state, assemble to form a helical complex encompassing the R and I sites in oriC (Fig. 25-10). DnaA has a higher affinity for R sites than I sites, and it binds R sites equally well in its ATP- or ADP-bound form. The I sites, which bind only the ATP-bound DnaA, allow discrimination between the active and inactive forms of DnaA. The tight right-handed wrapping of the DNA around this complex introduces a positive supercoil (see Chapter 24). The associated strain in the nearby DNA, combined with the binding of additional DnaA protein to the DUE region, leads to strand separation in the DUE. The complex formed at the replication origin also includes several DNA-binding proteins — and — that facilitate DNA bending.
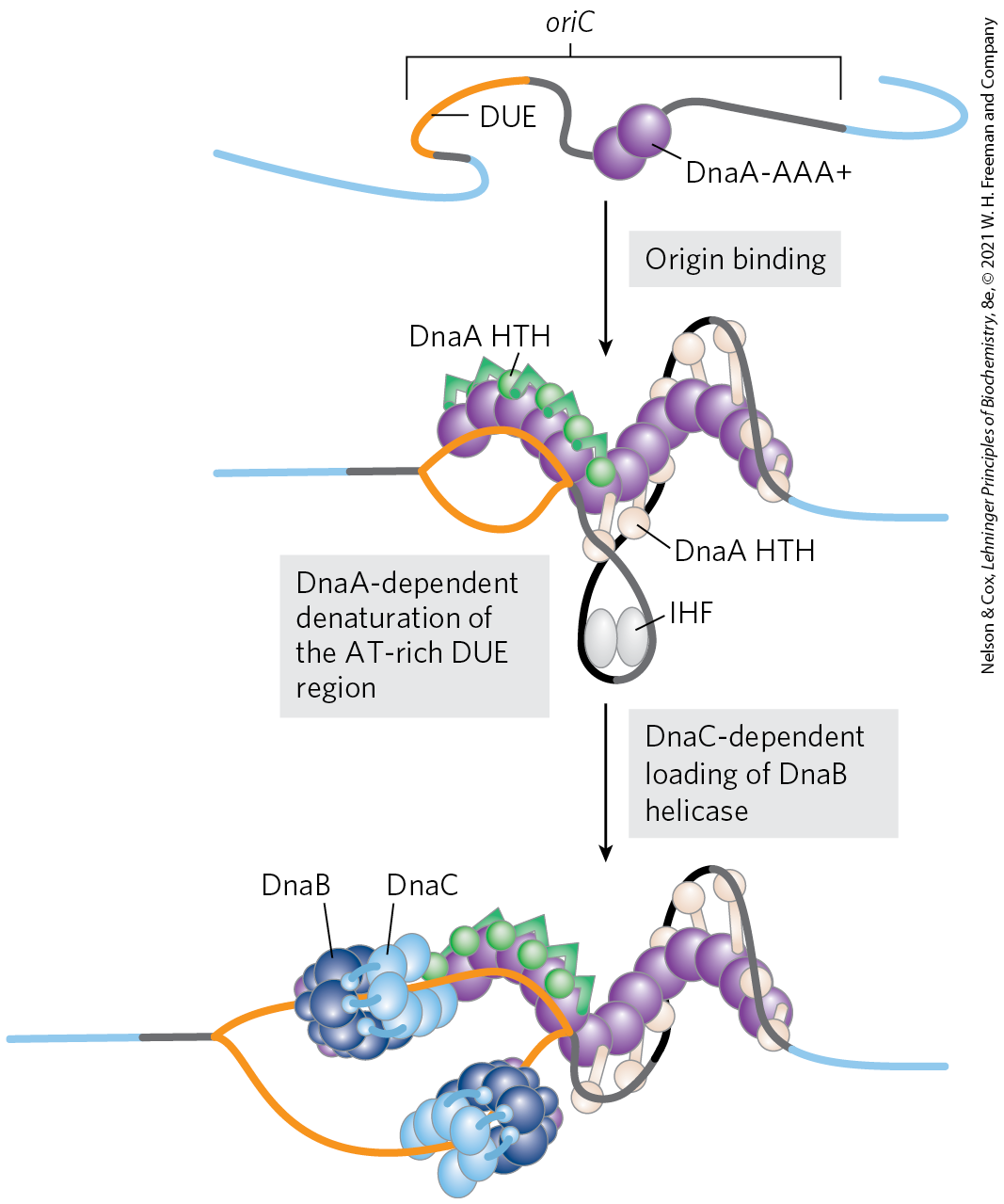
FIGURE 25-10 Model for initiation of replication at the E. coli origin, oriC. DnaA protein molecules bind initially at the five specific R sites. Upon ATP binding, additional DnaA molecules bind at the I sites in the origin, forming a right-handed helical complex and drawing in more DnaA molecules that continue the helix into the DUE region (see Fig. 25-9). The DNA is wrapped around this complex. DnaA molecules bound at the R and I sites bind the DNA with an HTH doman (referring to a DNA binding motif called helix-turn-helix). The -rich DUE region is denatured as a result of the strain imparted by the DnaA binding. Within the DUE, single-stranded DNA is bound by the ATPase domain of DnaA rather than by the HTH. Formation of the helical DnaA complex is facilitated by the proteins HU, IHF, and FIS. The detailed structural roles of these proteins are not known, but IHF may stabilize a transient DNA loop, as shown here. Hexamers of the DnaB protein bind to each strand, with the aid of DnaC protein. The DnaB helicase activity further unwinds the DNA in preparation for priming and DNA synthesis. [Information from J. P. Erzberger et al., Nat. Struct. Mol. Biol. 13:676, 2006.]
The DnaC protein, another , then loads the DnaB protein onto the separated DNA strands in the denatured region. A hexamer of DnaC, each subunit bound to ATP, forms a tight complex with the ring-shaped, hexameric DnaB helicase. This DnaC-DnaB interaction opens the DnaB ring, the process being aided by a further interaction between DnaB and DnaA. Two of the ring-shaped DnaB hexamers are loaded in the DUE, one onto each DNA strand. The ATP bound to DnaC is hydrolyzed, releasing the DnaC and leaving the DnaB bound to the DNA.
Loading of the DnaB helicase is the key step in replication initiation. As a replicative helicase, DnaB migrates along the single-stranded DNA in the direction, unwinding the DNA as it travels. The DnaB helicases loaded onto the two DNA strands thus travel in opposite directions, creating two potential replication forks. All other proteins at the replication fork are linked directly or indirectly to DnaB. The DNA polymerase III holoenzyme is linked through its subunits; additional DnaB interactions are described below. As replication begins and the DNA strands are separated at the fork, many molecules of single-stranded DNA–binding protein (SSB) bind to and stabilize the separated strands, and DNA gyrase (DNA topoisomerase II) relieves the topological stress induced ahead of the fork by the unwinding reaction.
Initiation is the only phase of DNA replication that is known to be regulated, and it is regulated such that replication occurs only once in each cell cycle. The mechanism of regulation is not yet entirely understood, but genetic and biochemical studies have provided insights into several separate regulatory mechanisms.
Once DNA polymerase III has been loaded onto the DNA, along with the β subunits (signaling completion of the initiation phase), the protein Hda binds to the β subunits and interacts with DnaA to stimulate hydrolysis of its bound ATP. Hda is yet another closely related to DnaA (its name is derived from homologous to DnaA). This ATP hydrolysis leads to disassembly of the DnaA complex at the origin. Slow release of ADP by DnaA and rebinding of ATP cycles the protein between its inactive (with bound ADP) and active (with bound ATP) forms on a time scale of 20 to 40 minutes.
The timing of replication initiation is affected by DNA methylation and interactions with the bacterial plasma membrane. The oriC DNA is methylated by the Dam methylase (Table 25-3), which methylates the position of adenine within the palindromic sequence GATC. (Dam is not a biochemical expletive; it stands for DNA adenine methylation.) The oriC region of E. coli is highly enriched in GATC sequences — it has 11 in its 245 bp; the average frequency of GATC in the E. coli chromosome as a whole is 1 in 256 bp.
Immediately after replication, the DNA is hemimethylated: the parent strands have methylated oriC sequences but the newly synthesized strands do not. The hemimethylated oriC sequences are now sequestered by interaction with the plasma membrane (the mechanism is unknown) and by binding of the protein SeqA. After a time, oriC is released from the plasma membrane, SeqA dissociates, and the DNA must be fully methylated by Dam methylase before it can again bind DnaA and initiate a new round of replication.
Elongation
The elongation phase of replication includes two distinct but related operations: leading strand synthesis and lagging strand synthesis. Several enzymes at the replication fork are important to the synthesis of both strands. Parent DNA is first unwound by DNA helicases, and the resulting topological stress is relieved by topoisomerases. Each separated strand is then stabilized by SSB. From this point, synthesis of leading and lagging strands is sharply different.
Leading strand synthesis, the more straightforward of the two, begins with the synthesis by primase (DnaG protein) of a short (10 to 60 nucleotide) RNA primer at the replication origin. DnaG interacts with DnaB helicase to carry out this reaction, and the primer is synthesized in the direction opposite to that in which the DnaB helicase is moving. In effect, the DnaB helicase moves along the strand that becomes the lagging strand in DNA synthesis; however, the first primer laid down in the first DnaG-DnaB interaction serves to prime leading strand DNA synthesis in the opposite direction. Deoxyribonucleotides are added to this primer by a DNA polymerase III complex linked to the DnaB helicase tethered to the opposite DNA strand. Leading strand synthesis then proceeds continuously, keeping pace with the unwinding of DNA at the replication fork.
Lagging strand synthesis, as we have noted, is accomplished in short Okazaki fragments (Fig. 25-11a). First, an RNA primer is synthesized by primase, and, as in leading strand synthesis, DNA polymerase III binds to the RNA primer and adds deoxyribonucleotides (Fig. 25-11b). On this level, the synthesis of each Okazaki fragment seems straightforward, but the details are quite complex. The complexity lies in the coordination of leading and lagging strand synthesis. Both strands are produced by a single asymmetric DNA polymerase III dimer; this is accomplished by looping the DNA of the lagging strand as shown in Figure 25-12, bringing together the two points of polymerization.
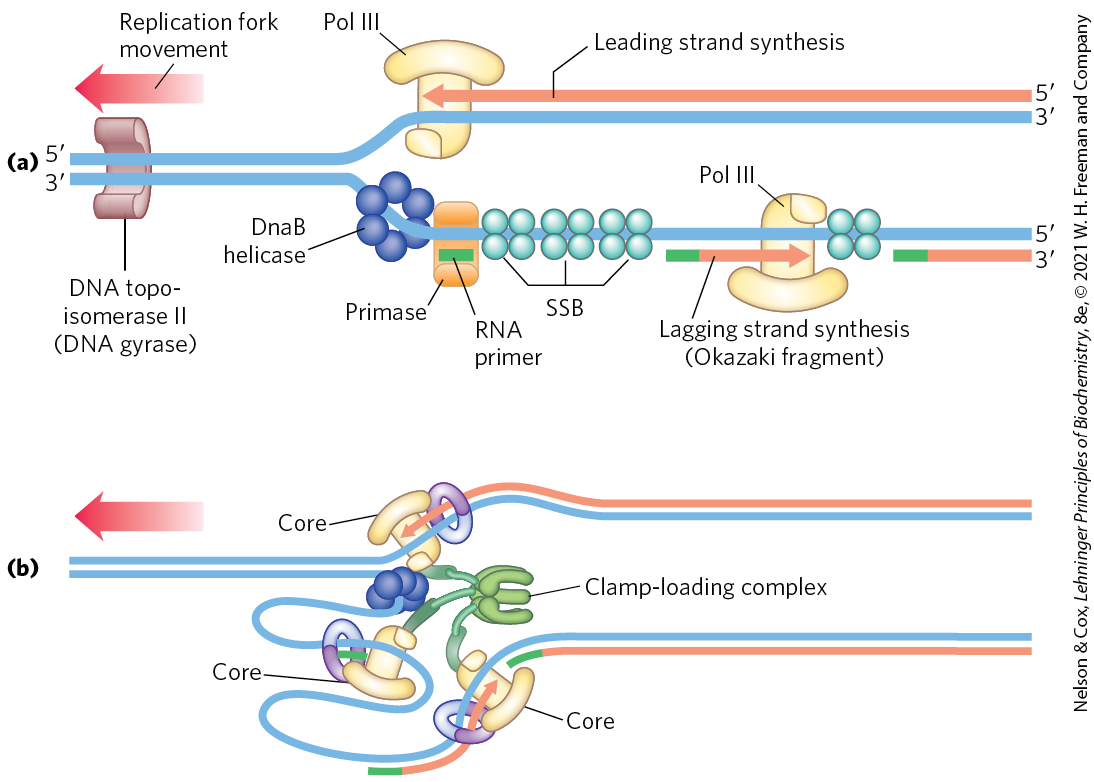
FIGURE 25-11 Synthesis of Okazaki fragments. (a) At intervals, primase synthesizes an RNA primer for a new Okazaki fragment. If we consider the two template strands as lying side by side, lagging strand synthesis formally proceeds in the opposite direction from fork movement. Each primer is extended by DNA polymerase III. DNA synthesis continues until the fragment extends as far as the primer of the previously added Okazaki fragment. A new primer is synthesized near the replication fork to begin the process again. (b) In the replisome complex, DNA synthesis on the leading and lagging strands is tightly coordinated. Each DNA polymerase III holoenzyme has three sets of core subunits (yellow), linked together with a single clamp-loading complex, so one or two Okazaki fragments can be synthesized simultaneously, along with the leading strand.
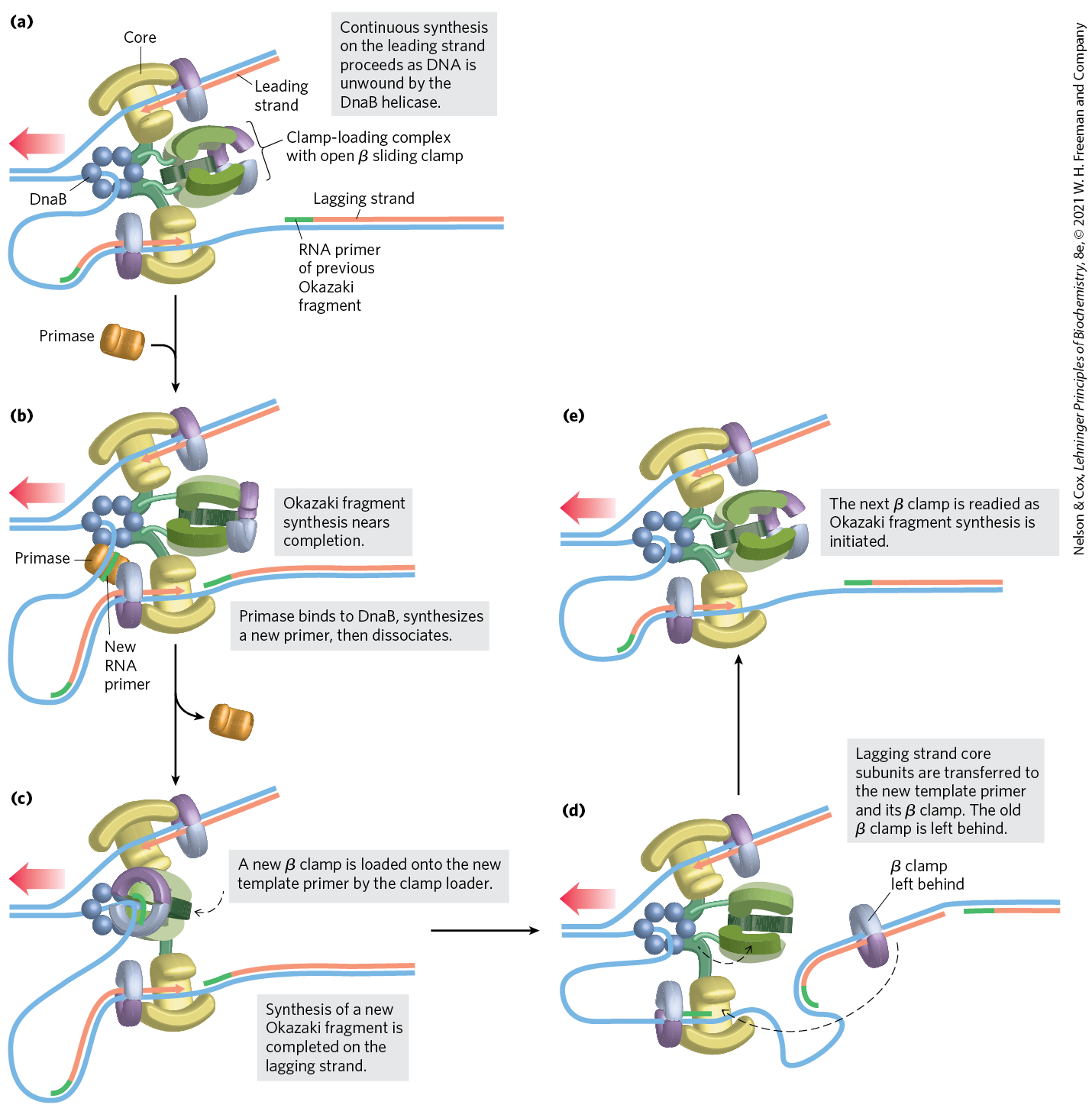
FIGURE 25-12 DNA synthesis on the leading and lagging strands. Events at the replication fork are coordinated by a single DNA polymerase III dimer, in an integrated complex with DnaB helicase. This figure shows the replication process already underway; (a) through (e) are discussed in the text. Only two sets of polymerase core subunits rather than three are shown, to more clearly illustrate the cycling on the lagging strand. The lagging strand is looped so that DNA synthesis proceeds steadily on both the leading and lagging strand templates at the same time. Red arrows indicate the end of the two new strands and the direction of DNA synthesis. An Okazaki fragment is being synthesized on the lagging strand. The subunit colors and the functions of the clamp-loading complex are explained in Figure 25-13.
The synthesis of Okazaki fragments on the lagging strand entails some elegant enzymatic choreography. DNA polymerase III uses one set of its core subunits (the core polymerase) to synthesize the leading strand continuously, while the other two sets of core subunits cycle from one Okazaki fragment to the next on the looped lagging strand. In vitro, a DNA polymerase III holoenzyme with only two sets of core subunits can synthesize both leading and lagging strands. However, a third set of core subunits increases the efficiency of lagging strand synthesis as well as the processivity of the overall replisome.
DnaB helicase, bound in front of DNA polymerase III, unwinds the DNA at the replication fork (Fig. 25-12a) as it travels along the lagging strand template in the direction. DnaG primase occasionally associates with DnaB helicase and synthesizes a short RNA primer (Fig. 25-12b). A new β sliding clamp is then positioned at the primer by the clamp-loading complex of DNA polymerase III (Fig. 25-12c). When synthesis of an Okazaki fragment has been completed, replication halts, and the core subunits of DNA polymerase III dissociate from their β sliding clamp (and from the completed Okazaki fragment) and associate with the new clamp (Fig. 25-12d, e). This initiates synthesis of a new Okazaki fragment. Two sets of core subunits may be engaged in the synthesis of two different Okazaki fragments at the same time. The proteins acting at the replication fork are summarized in Table 25-4.
| Protein | Number of subunits | Function | |
|---|---|---|---|
SSB |
75,600 |
4 |
Binding to single-stranded DNA |
Helicase (DnaB protein) |
300,000 |
6 |
DNA unwinding |
Primase (DnaG protein) |
60,000 |
1 |
RNA primer synthesis |
DNA polymerase III |
1,065,400 |
17 |
New strand elongation |
DNA polymerase I |
103,000 |
1 |
Filling of gaps; excision of primers |
DNA ligase |
74,000 |
1 |
Ligation |
DNA gyrase (DNA topoisomerase II) |
400,000 |
4 |
Supercoiling |
The clamp-loading complex of DNA polymerase III, consisting of parts of the three subunits along with the and subunits, is also an . This complex binds to ATP and to the new β sliding clamp. The binding imparts strain on the dimeric clamp, opening up the ring at one subunit interface (Fig. 25-13). The newly primed lagging strand is slipped into the ring through the resulting break. The clamp loader then hydrolyzes ATP, releasing the β sliding clamp and allowing it to close around the DNA.
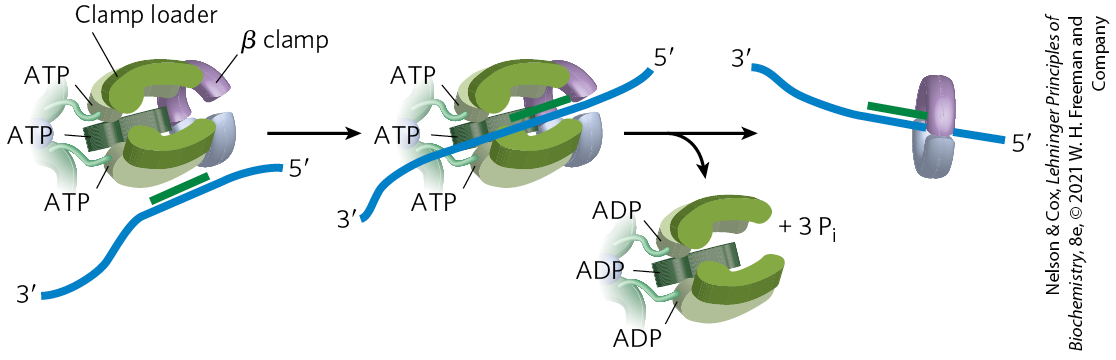
FIGURE 25-13 The DNA polymerase III clamp loader. The five subunits of the clamp-loading complex are the and subunits and the amino-terminal domain of each of the three subunits (see Fig. 25-8). The complex binds to three molecules of ATP and to a dimeric β clamp. This binding forces the β clamp open at one of its two subunit interfaces. Hydrolysis of the bound ATP allows the β clamp to close again around the DNA.
The replisome promotes rapid DNA synthesis, adding ~1,000 to 2,000 nucleotides to each strand (leading and lagging). Once an Okazaki fragment has been completed, its RNA primer is removed by DNA polymerase I or RNase H1 and replaced with DNA by the polymerase; the remaining nick is sealed by DNA ligase (Fig. 25-14).
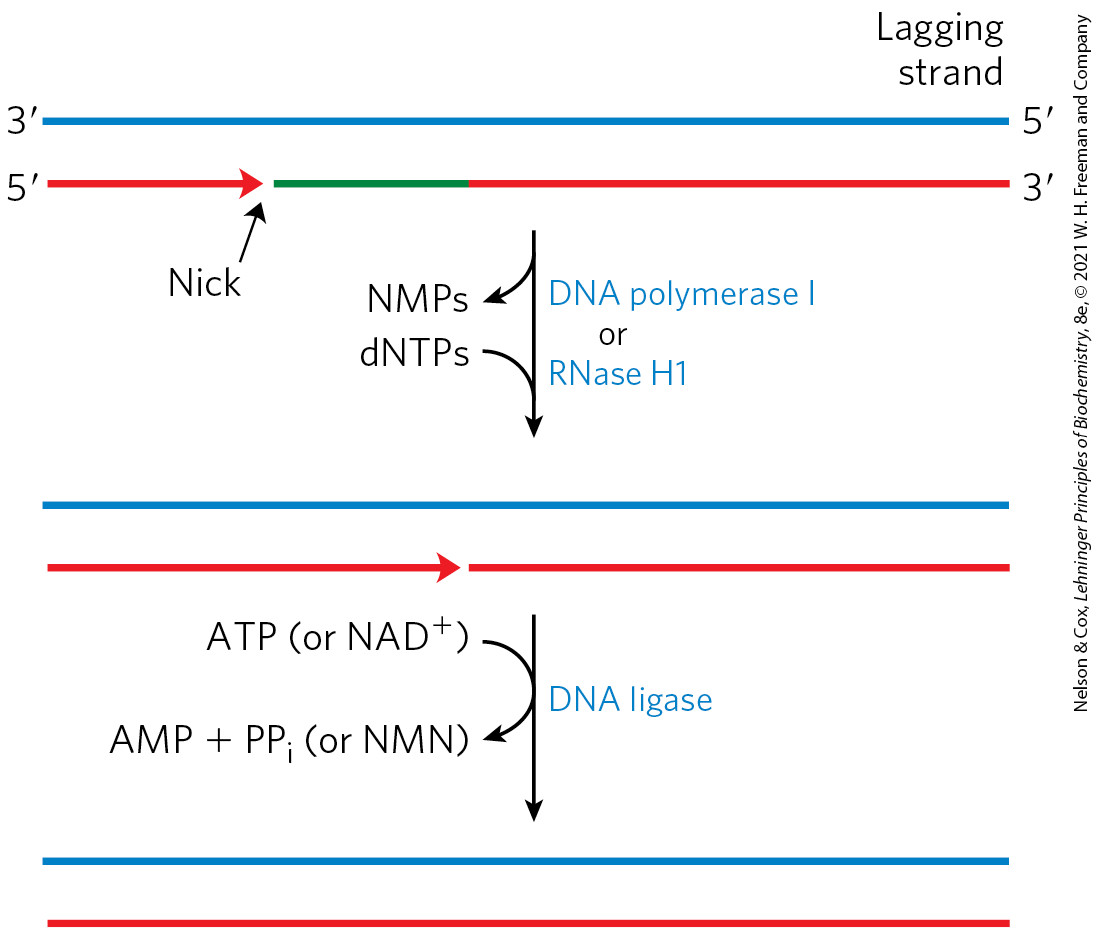
FIGURE 25-14 Final steps in the synthesis of lagging strand segments. RNA primers in the lagging strand are removed by the exonuclease activity of DNA polymerase I or RNase H1, and then replaced with DNA by DNA polymerase I. The remaining nick is sealed by DNA ligase. The role of ATP or is shown in Figure 25-15.
DNA ligase catalyzes the formation of a phosphodiester bond between a hydroxyl at the end of one DNA strand and a phosphate at the end of another strand. The phosphate must be activated by adenylylation. DNA ligases isolated from viruses and eukaryotes use ATP for this purpose. DNA ligases from bacteria are unusual in that many use — a cofactor that usually functions in hydride transfer reactions (see Fig. 13-24) — as the source of the AMP activating group (Fig. 25-15). DNA ligase is another enzyme of DNA metabolism that has become an important reagent in recombinant DNA experiments (see Fig. 9-1).
FIGURE 25-15 Mechanism of the DNA ligase reaction. In each of the three steps, one phosphodiester bond is formed at the expense of another. Steps and lead to activation of the phosphate in the nick. An AMP group is transferred first to a Lys residue on the enzyme and then to the phosphate in the nick. In step , the -hydroxyl group attacks this phosphate and displaces AMP, producing a phosphodiester bond to seal the nick. In the E. coli DNA ligase reaction, AMP is derived from . The DNA ligases isolated from some viral and eukaryotic sources use ATP rather than and they release pyrophosphate rather than nicotinamide mononucleotide (NMN) in step .
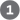 and
and  lead to activation of the phosphate in the nick. An AMP group is transferred first to a Lys residue on the enzyme and then to the phosphate in the nick. In step
lead to activation of the phosphate in the nick. An AMP group is transferred first to a Lys residue on the enzyme and then to the phosphate in the nick. In step  , the -hydroxyl group attacks this phosphate and displaces AMP, producing a phosphodiester bond to seal the nick. In the E. coli DNA ligase reaction, AMP is derived from . The DNA ligases isolated from some viral and eukaryotic sources use ATP rather than and they release pyrophosphate rather than nicotinamide mononucleotide (NMN) in step
, the -hydroxyl group attacks this phosphate and displaces AMP, producing a phosphodiester bond to seal the nick. In the E. coli DNA ligase reaction, AMP is derived from . The DNA ligases isolated from some viral and eukaryotic sources use ATP rather than and they release pyrophosphate rather than nicotinamide mononucleotide (NMN) in step Termination
Eventually, the two replication forks of the circular E. coli chromosome meet at a terminus region containing multiple copies of a 20 bp sequence called Ter (Fig. 25-16). The Ter sequences are arranged on the chromosome to create a trap that a replication fork can enter but cannot leave. The Ter sequences function as binding sites for the protein Tus (terminus utilization substance). The Tus-Ter complex can arrest a replication fork from only one direction. Only one Tus-Ter complex functions per replication cycle — the complex first encountered by either replication fork. Given that opposing replication forks generally halt when they collide, Ter sequences would not seem to be essential, but they may prevent overreplication by one fork in the event that the other is delayed or halted by an encounter with DNA damage or some other obstacle.
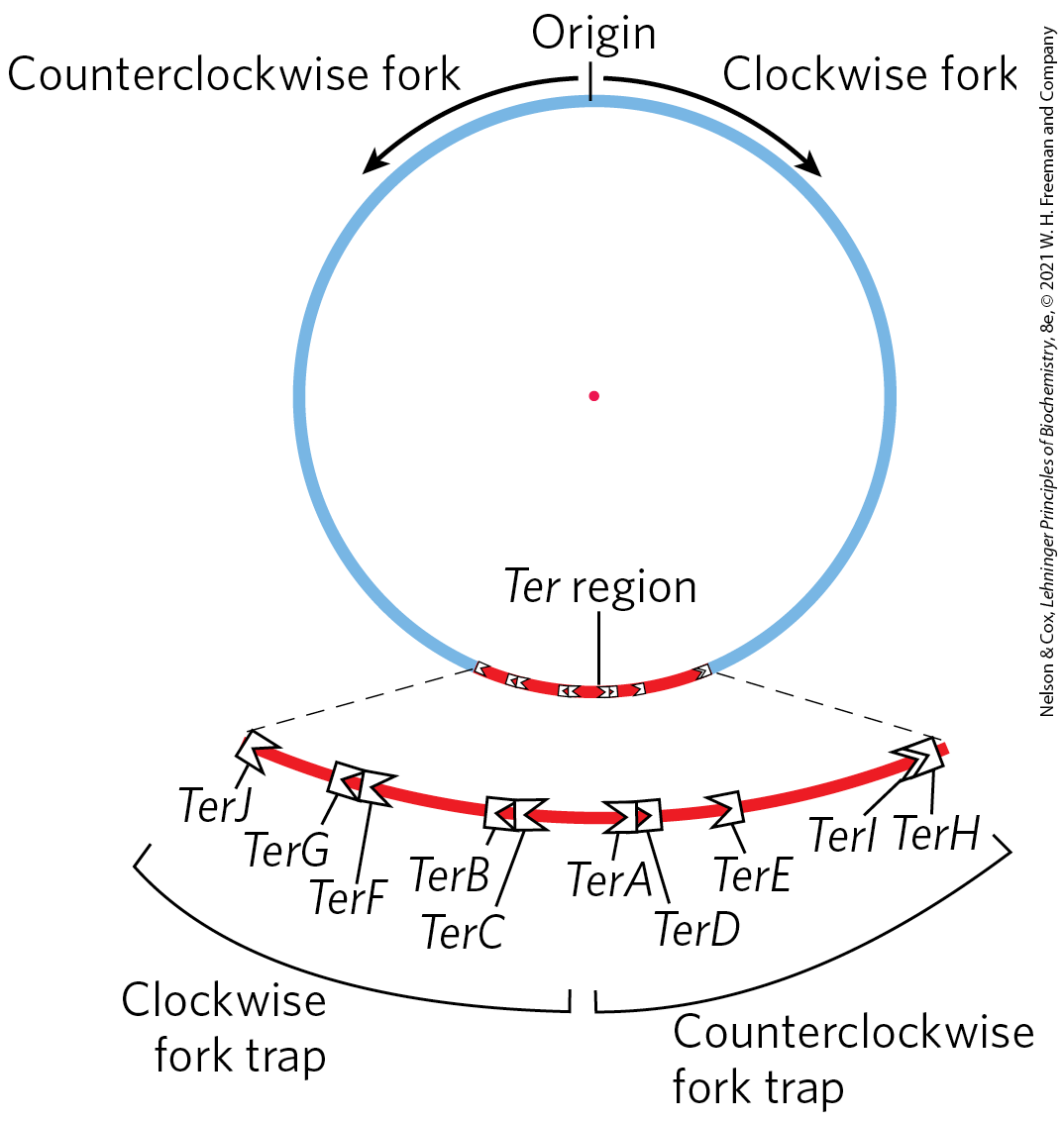
FIGURE 25-16 Termination of chromosome replication in E. coli. The Ter sequences (TerA through TerJ) are positioned on the chromosome in two clusters with opposite orientations. The overall Ter region encompasses about 9% of the circular chromosome.
So, when either replication fork encounters a functional Tus-Ter complex, it halts; the other fork halts when it meets the first (arrested) fork. The final few hundred base pairs of DNA between these large protein complexes are then replicated (by an as yet unknown mechanism), completing two topologically interlinked (catenated) circular chromosomes (Fig. 25-17). DNA circles linked in this way are known as catenanes. Separation of the catenated circles in E. coli requires topoisomerase IV (a type II topoisomerase). The separated chromosomes then segregate into daughter cells at cell division. The terminal phase of replication of other circular chromosomes, including many of the DNA viruses that infect eukaryotic cells, is similar.
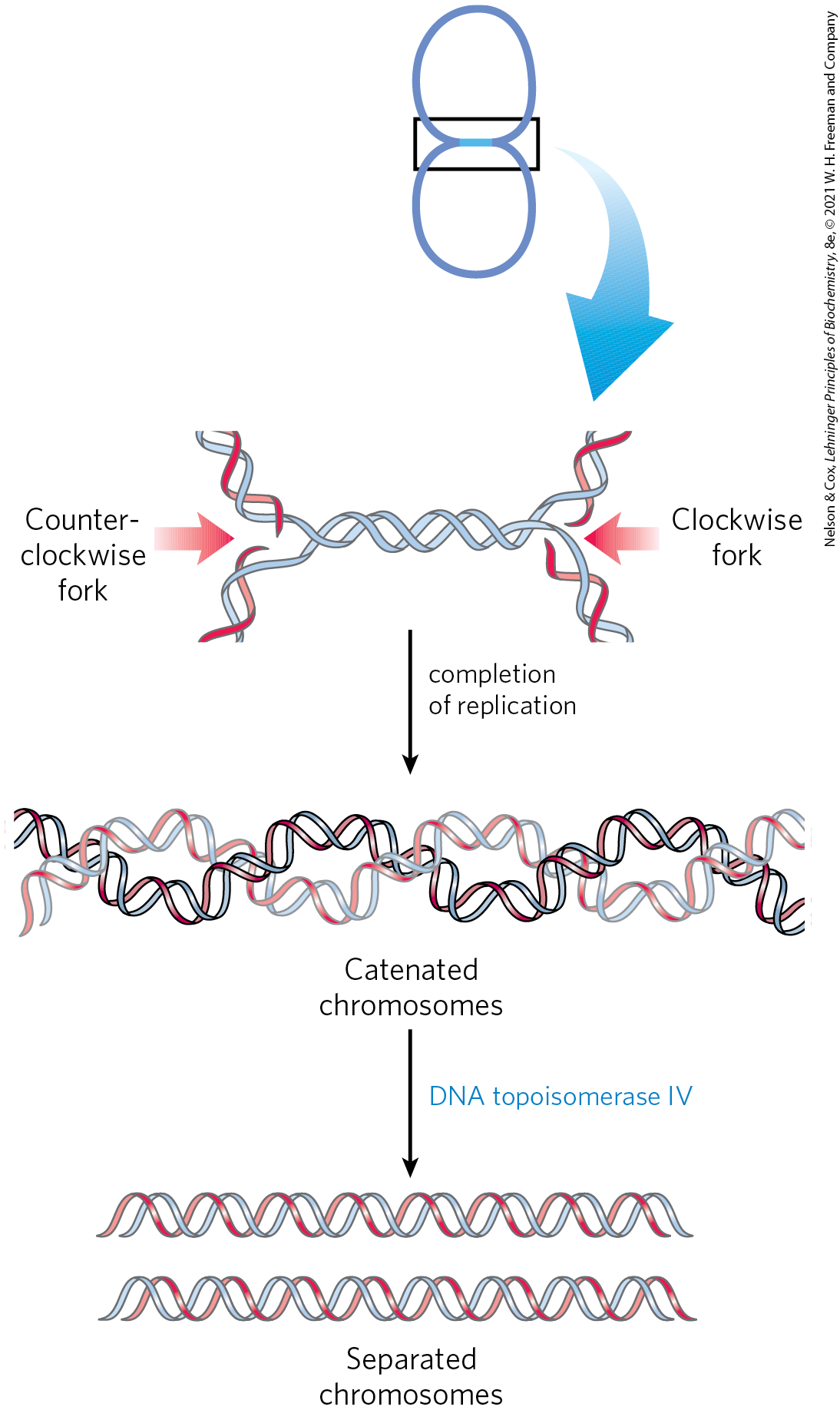
FIGURE 25-17 Role of topoisomerases in replication termination. Replication of the DNA separating opposing replication forks leaves the completed chromosomes joined as catenanes, or topologically interlinked circles. The circles are not covalently linked, but because they are interwound and each is covalently closed, they cannot be separated — except by the action of topoisomerases. In E. coli, a type II topoisomerase known as DNA topoisomerase IV plays the primary role in separating catenated chromosomes, transiently breaking both DNA strands of one chromosome and allowing the other chromosome to pass through the break.
Replication in Eukaryotic Cells Is Similar but More Complex
The DNA molecules in eukaryotic cells are considerably larger than those in bacteria and are organized into complex nucleoprotein structures (chromatin; p. 898). The essential features of DNA replication are the same in eukaryotes and bacteria, and many of the protein complexes are functionally and structurally conserved. However, eukaryotic replication is regulated and coordinated with the cell cycle, and must function within the complexities of chromatin structure.
Origins of replication have a well-characterized structure in some lower eukaryotes, but they are much less defined in higher eukaryotes. In both cases, replication begins in short nucleosome-free regions. Yeast (S. cerevisiae) has about 400 defined replication origins called autonomously replicating sequences (ARSs), or replicators. Yeast replicators span ∼150 bp and contain several essential, conserved sequences. There are about 30,000 to 50,000 replication origins in human chromosomes. The replication origins of vertebrates in general may be defined by some aspect of DNA secondary structure, as yet unknown.
Regulation ensures that all cellular DNA is replicated once per cell cycle. Much of this regulation involves proteins called cyclins and the cyclin-dependent kinases (CDKs) with which they form complexes (see Section 12.8). The cyclins are rapidly destroyed by ubiquitin-dependent proteolysis at the end of the M phase (mitosis), and the absence of cyclins allows the establishment of prereplicative complexes (pre-RCs) on replication initiation sites. In rapidly growing cells, the pre-RC forms at the end of M phase. In slow-growing cells, it does not form until the end of G1. Formation of the pre-RC renders the cell competent for replication, an event sometimes called licensing.
As in bacteria, the key event in the initiation of replication in all eukaryotes is the loading of the replicative helicase, a heterohexameric complex of minichromosome maintenance (MCM) proteins (MCM2 to MCM7). The ring-shaped MCM2–7 helicase functions in some ways like the bacterial DnaB helicase, although it translocates along the leading strand template. It is loaded onto the DNA in steps (Fig. 25-18). The origin is recognized and bound first by another six-protein complex, called ORC (origin recognition complex), followed by the protein CDC6 (cell division cycle), which recruits CDT1 (CDC10-dependent transcript 1). Together, they facilitate the loading of two inactive MCM2–7 complexes (the pre-RC). The ORC-CDC6 complex and CTD1 dissociate, leaving behind the pre-RC. ORC has five domains among its subunits and is functionally analogous to the bacterial DnaA. The yeast CDC6 is yet another that forms a complex with the ORC subunits. Following pre-RC formation, another set of proteins, CDC45 and the GINS, bind to and activate the MCM2–7 helicase, triggering DNA denaturation. (GINS refers to the first letters of the numbers 5-1-2-3 in Japanese, go-ichi-ni-san, providing a somewhat cryptic callout to the four protein subunits of the complex: SLD5, PSF1, PSF2, and PSF3.) The replication proteins then bind to form a replisome, and bidirectional replication begins.
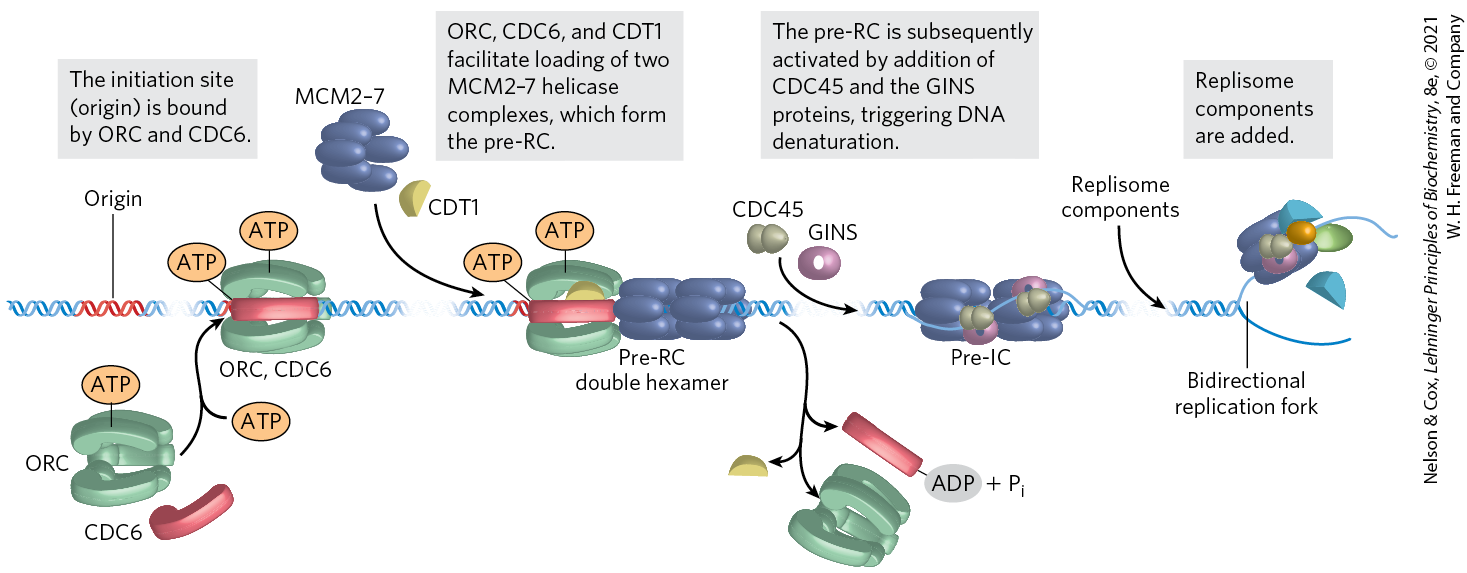
FIGURE 25-18 Assembly of a prereplicative complex at a eukaryotic replication origin. The initiation site (origin) is bound by ORC, CDC6, and CDT1. These proteins, many of them , promote loading of two MCM2–7 helicase complexes, in a reaction analogous to the loading of the bacterial DnaB helicase by DnaC protein. The two loaded but inactive MCM2–7 complexes comprise the prereplicative complex, or pre-RC. The pre-RC is subsequently activated by addition of CDC45 and the GINS proteins, followed by addition of the replisome components. [Information from M. W. Parker et al., Crit. Rev. Biochem. Mol. Biol. 52:107, 2017.]
Commitment to replication requires the synthesis and activity of S-phase cyclin-CDK complexes (such as the cyclin E–CDK2 complex; see Fig. 12-36) and CDC7-DBF4. Both types of complexes help to activate replication by binding to and phosphorylating several subunits of the pre-RC. Other cyclins and CDKs function to inhibit the formation of more pre-RC complexes once replication has been initiated. For example, CDK2 binds to cyclin A as cyclin E levels decline during S phase, inhibiting CDK2 and preventing the licensing of additional pre-RC complexes.
The rate of movement of the replication fork in eukaryotes (∼50 nucleotides/s) is only one-twentieth that observed in E. coli. At this rate, replication of an average human chromosome proceeding from a single origin would take more than 500 hours, making the requirement for many origins evident.
Like bacteria, eukaryotes have several types of DNA polymerases. Some have been linked to particular functions, such as the replication of mitochondrial DNA. The replication of nuclear chromosomes primarily involves three multisubunit DNA polymerases. The highly processive DNA polymerase ε synthesizes the leading strand, and DNA polymerase synthesizes the lagging strand. Both enzymes have proofreading exonuclease activities. DNA polymerase α, a DNA polymerase/primase, synthesizes RNA primers and also extends them by about 10 nucleotides of DNA to initiate synthesis of each Okazaki fragment on the lagging strand. One subunit of DNA polymerase α has a primase activity, and the largest subunit contains the polymerization activity. However, this polymerase has no proofreading exonuclease activity, making it unsuitable for high-fidelity DNA replication.
DNA polymerases ε and δ are associated with and stimulated by proliferating cell nuclear antigen (PCNA; ), a protein found in large amounts in the nuclei of proliferating cells. The three-dimensional structure of PCNA is remarkably similar to that of the β subunit of E. coli DNA polymerase III (Fig. 25-8b), although primary sequence homology is not evident. PCNA has a function analogous to that of the β subunit, forming a circular clamp that enhances the processivity of the two polymerases.
Two other protein complexes also function in eukaryotic DNA replication. RPA (replication protein A) is a single-stranded DNA–binding protein, equivalent in function to the E. coli SSB protein. RFC (replication factor C) is a clamp loader for PCNA and facilitates the assembly of active replication complexes. The subunits of the RFC complex have significant sequence similarity to the subunits of the bacterial clamp-loading complex.
Termination of replication on linear eukaryotic chromosomes occurs when replication forks operating from nearby origins converge. As in bacteria, there are successive steps of final replication, replisome dissociation, and decatenation of the DNA products. All of the steps are mediated by additional protein complexes, with some parts of the process still undefined.
Viral DNA Polymerases Provide Targets for Antiviral Therapy
Many DNA viruses encode their own DNA polymerases, and some of these have become targets for pharmaceuticals. For example, the DNA polymerase of the herpes simplex virus is inhibited by acyclovir, a compound developed by Gertrude Elion and George Hitchings (p. 836). Acyclovir consists of guanine attached to an incomplete ribose ring.
 Many DNA viruses encode their own DNA polymerases, and some of these have become targets for pharmaceuticals. For example, the DNA polymerase of the herpes simplex virus is inhibited by acyclovir, a compound developed by Gertrude Elion and George Hitchings (
Many DNA viruses encode their own DNA polymerases, and some of these have become targets for pharmaceuticals. For example, the DNA polymerase of the herpes simplex virus is inhibited by acyclovir, a compound developed by Gertrude Elion and George Hitchings (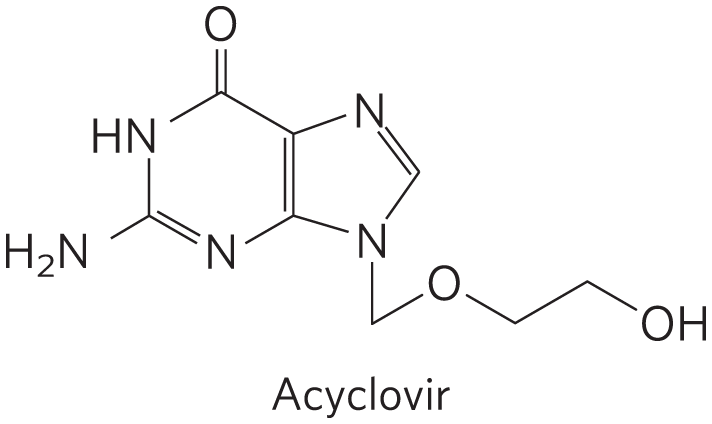
It is phosphorylated by a virally encoded thymidine kinase; acyclovir binds to this viral enzyme with an affinity 200-fold greater than its binding to the cellular thymidine kinase. This ensures that phosphorylation occurs mainly in virus-infected cells. Cellular kinases convert the resulting acyclo-GMP to acyclo-GTP, which is both an inhibitor and a substrate of DNA polymerases; acyclo-GTP competitively inhibits the herpes DNA polymerase more strongly than cellular DNA polymerases. Because it lacks a hydroxyl, acyclo-GTP also acts as a chain terminator when incorporated into DNA. Thus viral replication is inhibited at several steps.

SUMMARY 25.1 DNA Replication
- Replication of DNA follows a set of universal rules. Replication is semiconservative, each strand acting as template for a new daughter strand. It is carried out in three identifiable phases: initiation, elongation, and termination. The process starts at a single origin in bacteria and usually proceeds bidirectionally. DNA is synthesized in the direction by DNA polymerases. At the replication fork, the leading strand is synthesized continuously in the same direction as replication fork movement; the lagging strand is synthesized discontinuously as Okazaki fragments, which are subsequently ligated.
- Nucleases are enzymes that degrade DNA. Endonucleases cleave within a DNA polymer; exonucleases degrade DNA from the end of one strand (either or ).
- DNA polymerases are complex enzymes that synthesize DNA, and often possess additional activities, including exonuclease functions.
- DNA is replicated with very high fidelity. Accuracy is maintained by (1) base selection by the polymerase, (2) a proofreading exonuclease activity that is part of many DNA polymerases, and (3) specific repair systems for mismatches left behind after replication.
- Most cells have several DNA polymerases. In E. coli, DNA polymerase III is the primary replication enzyme. DNA polymerase I is responsible for special functions during replication, recombination, and repair.
- Replication requires an array of enzymes and protein factors in addition to DNA polymerases. Many of these proteins belong to the family.
- Replication initiation occurs when replicative helicases are loaded onto replication origins in stepwise fashion. Elongation is achieved by an active replisome — a supramolecular complex of nucleic acids and many proteins, including polymerases. Termination occurs when replisomes proceeding in opposite directions converge. It requires decatenation of the replication products when replication is complete.
- The major replicative DNA polymerases in eukaryotes are DNA polymerases ε and δ. DNA polymerase α synthesizes primers.
- Viral DNA replication is a drug target.
 Replication of DNA follows a set of universal rules. Replication is semiconservative, each strand acting as template for a new daughter strand. It is carried out in three identifiable phases: initiation, elongation, and termination. The process starts at a single origin in bacteria and usually proceeds bidirectionally. DNA is synthesized in the direction by DNA polymerases. At the replication fork, the leading strand is synthesized continuously in the same direction as replication fork movement; the lagging strand is synthesized discontinuously as Okazaki fragments, which are subsequently ligated.
Replication of DNA follows a set of universal rules. Replication is semiconservative, each strand acting as template for a new daughter strand. It is carried out in three identifiable phases: initiation, elongation, and termination. The process starts at a single origin in bacteria and usually proceeds bidirectionally. DNA is synthesized in the direction by DNA polymerases. At the replication fork, the leading strand is synthesized continuously in the same direction as replication fork movement; the lagging strand is synthesized discontinuously as Okazaki fragments, which are subsequently ligated.