12.8 Regulation of the Cell Cycle by Protein Kinases
One of the most dramatic manifestations of signaling pathways is the regulation of the eukaryotic cell cycle. During embryonic growth and later development, cell division occurs in virtually every tissue. In the adult organism, cells of most tissues stop dividing, becoming quiescent. A cell’s “decision” to divide or not is of crucial importance to the organism. When the regulatory mechanisms that limit cell division are defective and cells undergo unregulated division, the result is catastrophic — cancer. Proper cell division requires a precisely ordered sequence of biochemical events that assures every daughter cell a full complement of the molecules required for life. Investigations into the control of cell division in diverse eukaryotic cells have revealed universal regulatory mechanisms. Signaling mechanisms much like those discussed above are central in determining whether and when a cell undergoes cell division, and they also ensure orderly passage through the stages of the cell cycle.
 When the regulatory mechanisms that limit cell division are defective and cells undergo unregulated division, the result is catastrophic — cancer. Proper cell division requires a precisely ordered sequence of biochemical events that assures every daughter cell a full complement of the molecules required for life. Investigations into the control of cell division in diverse eukaryotic cells have revealed universal regulatory mechanisms. Signaling mechanisms much like those discussed above are central in determining whether and when a cell undergoes cell division, and they also ensure orderly passage through the stages of the cell cycle.
When the regulatory mechanisms that limit cell division are defective and cells undergo unregulated division, the result is catastrophic — cancer. Proper cell division requires a precisely ordered sequence of biochemical events that assures every daughter cell a full complement of the molecules required for life. Investigations into the control of cell division in diverse eukaryotic cells have revealed universal regulatory mechanisms. Signaling mechanisms much like those discussed above are central in determining whether and when a cell undergoes cell division, and they also ensure orderly passage through the stages of the cell cycle.The Cell Cycle Has Four Stages
Cell division accompanying mitosis in eukaryotes occurs in four well-defined stages (Fig. 12-35). In the S (synthesis) phase, the DNA is replicated to produce copies for both daughter cells. In the G2 phase (G indicates the gap between divisions), new proteins are synthesized and the cell approximately doubles in size. In the m phase (mitosis), the maternal nuclear envelope breaks down, paired chromosomes are pulled to opposite poles of the cell, each set of daughter chromosomes is surrounded by a newly formed nuclear envelope, and cytokinesis pinches the cell in half, producing two daughter cells (see Fig. 24-22). In embryonic or rapidly proliferating tissue, each daughter cell divides again, but only after a waiting period (G1). In animal cells in the laboratory, the entire process takes about 24 hours.
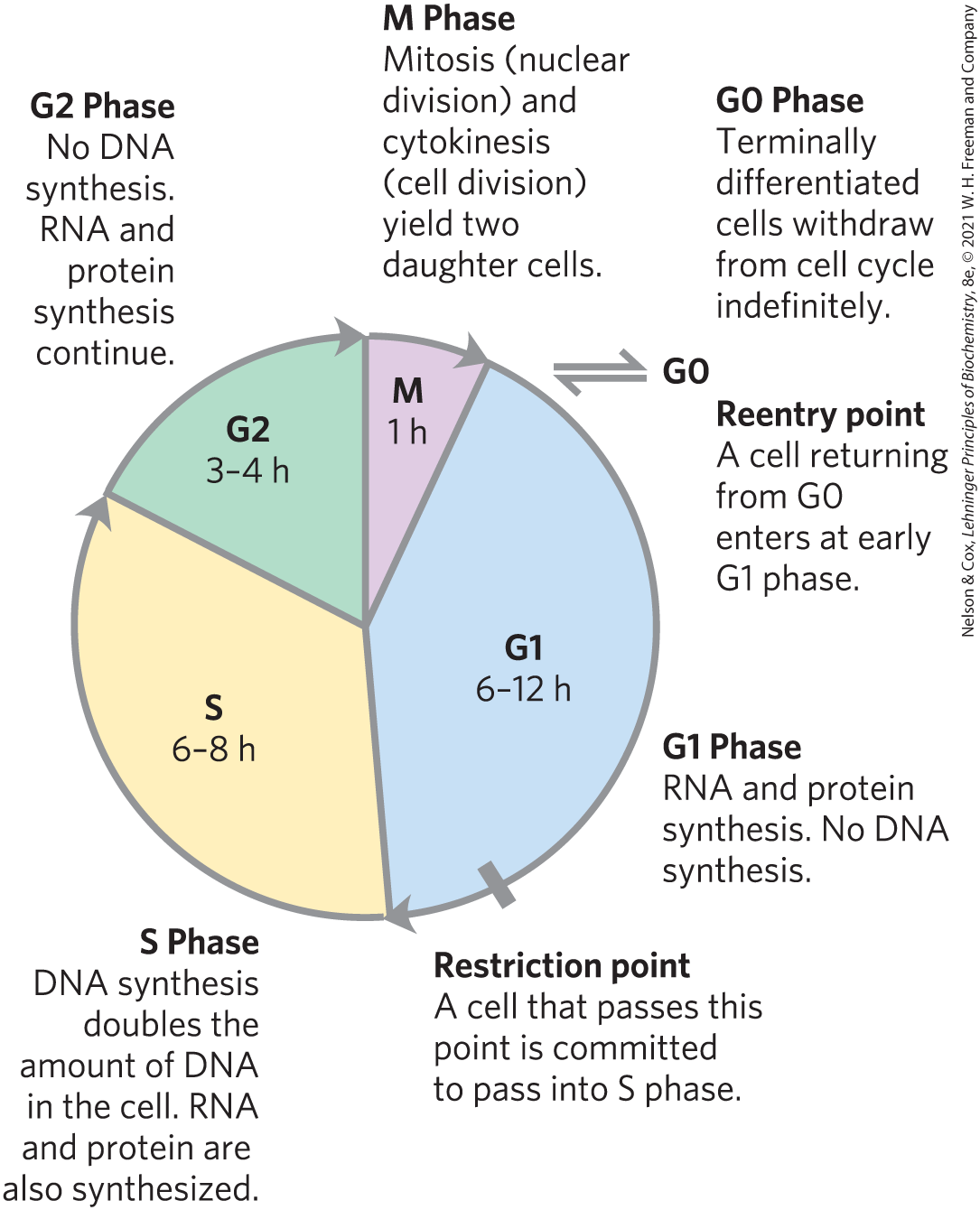
FIGURE 12-35 The eukaryotic cell cycle. The durations (in hours) of the four stages vary, but those shown are typical.
After passing through mitosis and into G1, a cell either continues through another division or ceases to divide, entering a quiescent phase (G0) that may last hours, days, or the lifetime of the cell. When a cell in G0 begins to divide again, it reenters the division cycle through the G1 phase. Differentiated cells such as hepatocytes or adipocytes have acquired their specialized function and form; they remain in the G0 phase. Stem cells retain their potential to divide and to differentiate into any of a number of cell types.
Levels of Cyclin-Dependent Protein Kinases Oscillate
The timing of the cell cycle is controlled by a family of protein kinases with activities that change in response to cellular signals. By phosphorylating specific proteins at precisely timed intervals, these protein kinases orchestrate the metabolic activities of the cell to produce orderly cell division. The kinases are heterodimers with a regulatory subunit, a cyclin, and a catalytic subunit, a cyclin-dependent protein kinase (CDK). In the absence of the cyclin, the catalytic subunit is virtually inactive. When the cyclin binds, the catalytic site opens up, a residue essential to catalysis becomes accessible, and the protein kinase activity of the catalytic subunit increases 10,000-fold. Animal cells have at least 10 different cyclins (designated A, B, and so forth) and at least 8 CDKs (CDK1 through CDK8), which act in various combinations at specific points in the cell cycle.
 The timing of the cell cycle is controlled by a family of protein kinases with activities that change in response to cellular signals. By phosphorylating specific proteins at precisely timed intervals, these protein kinases orchestrate the metabolic activities of the cell to produce orderly cell division. The kinases are heterodimers with a regulatory subunit, a
The timing of the cell cycle is controlled by a family of protein kinases with activities that change in response to cellular signals. By phosphorylating specific proteins at precisely timed intervals, these protein kinases orchestrate the metabolic activities of the cell to produce orderly cell division. The kinases are heterodimers with a regulatory subunit, a In a population of animal cells undergoing synchronous division, some CDK activities show striking oscillations (Fig. 12-36). These oscillations are the result of four mechanisms for regulating CDK activity: phosphorylation or dephosphorylation of the CDK, controlled degradation of the cyclin subunit, periodic synthesis of CDKs and cyclins, and the action of specific CDK-inhibiting proteins. The precisely timed activation and inactivation of a series of CDKs produces signals serving as a master clock that orchestrates the events in normal cell division and ensures that one stage is completed before the next begins.
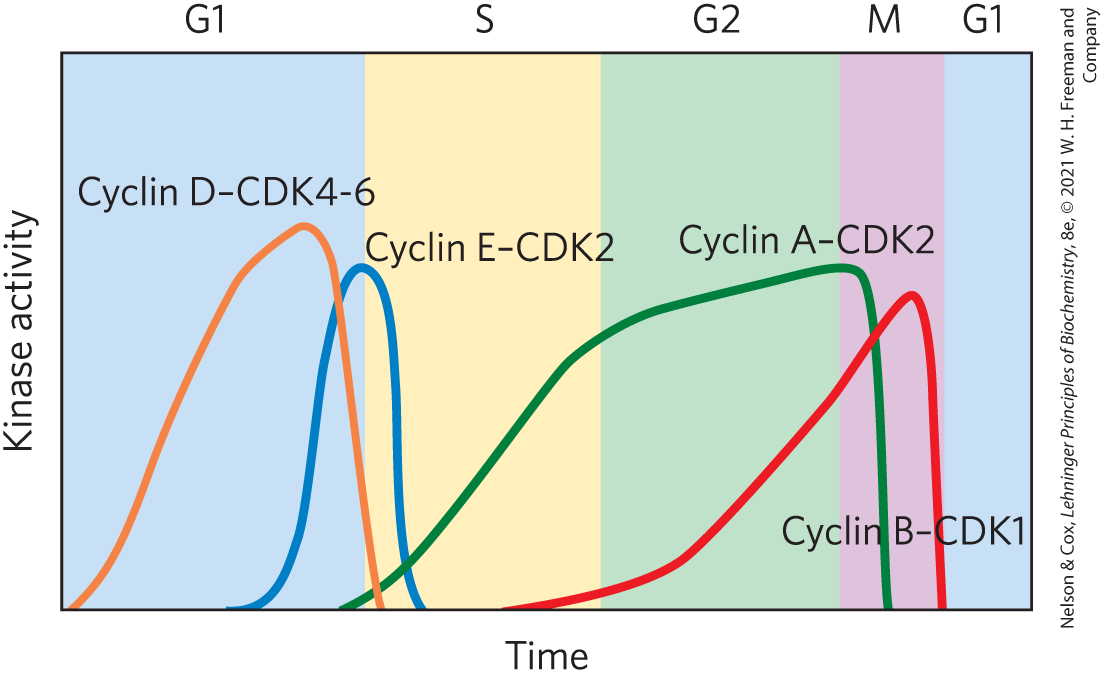
FIGURE 12-36 Variations in the activities of specific CDKs during the cell cycle in animals. Early in G1, the activity of cyclin D–CDK4-6 rises slowly, then drops sharply as G1 ends. Near the end of G1, cyclin E–CDK2 activity rises and peaks near the G1 phase–S phase boundary, when the active enzyme triggers synthesis of enzymes required for DNA synthesis (see Fig. 12-40). Cyclin A–CDK2 activity rises during the S and G2 phases, then drops sharply in the M phase, as cyclin B–CDK1 peaks. [Data from P. Icard et al., Trends Biochem. Sci. 44:490, 2019, Fig. 3.]
CDKs Are Regulated by Phosphorylation, Cyclin Degradation, Growth Factors, and Specific Inhibitors
The activity of a CDK is strikingly affected by phosphorylation and dephosphorylation of two specific residues in the protein (Fig. 12-37). Phosphorylation of of CDK2 stabilizes a conformation in which an autoinhibitory “T loop” is moved away from the substrate-binding cleft in the kinase, opening it to bind protein substrates. Dephosphorylation of - of CDK2 removes a negative charge that blocks ATP from approaching its binding site. This mechanism for activating a CDK is self-reinforcing; the enzyme (PTP) that dephosphorylates - is itself a substrate for the CDK and is activated by phosphorylation. The combination of these factors activates the CDK manyfold, allowing it to phosphorylate downstream protein targets required for progression of the cell cycle (Fig. 12-38a).
 - of CDK2 removes a negative charge that blocks ATP from approaching its binding site. This mechanism for activating a CDK is self-reinforcing; the enzyme (PTP) that dephosphorylates
- of CDK2 removes a negative charge that blocks ATP from approaching its binding site. This mechanism for activating a CDK is self-reinforcing; the enzyme (PTP) that dephosphorylates 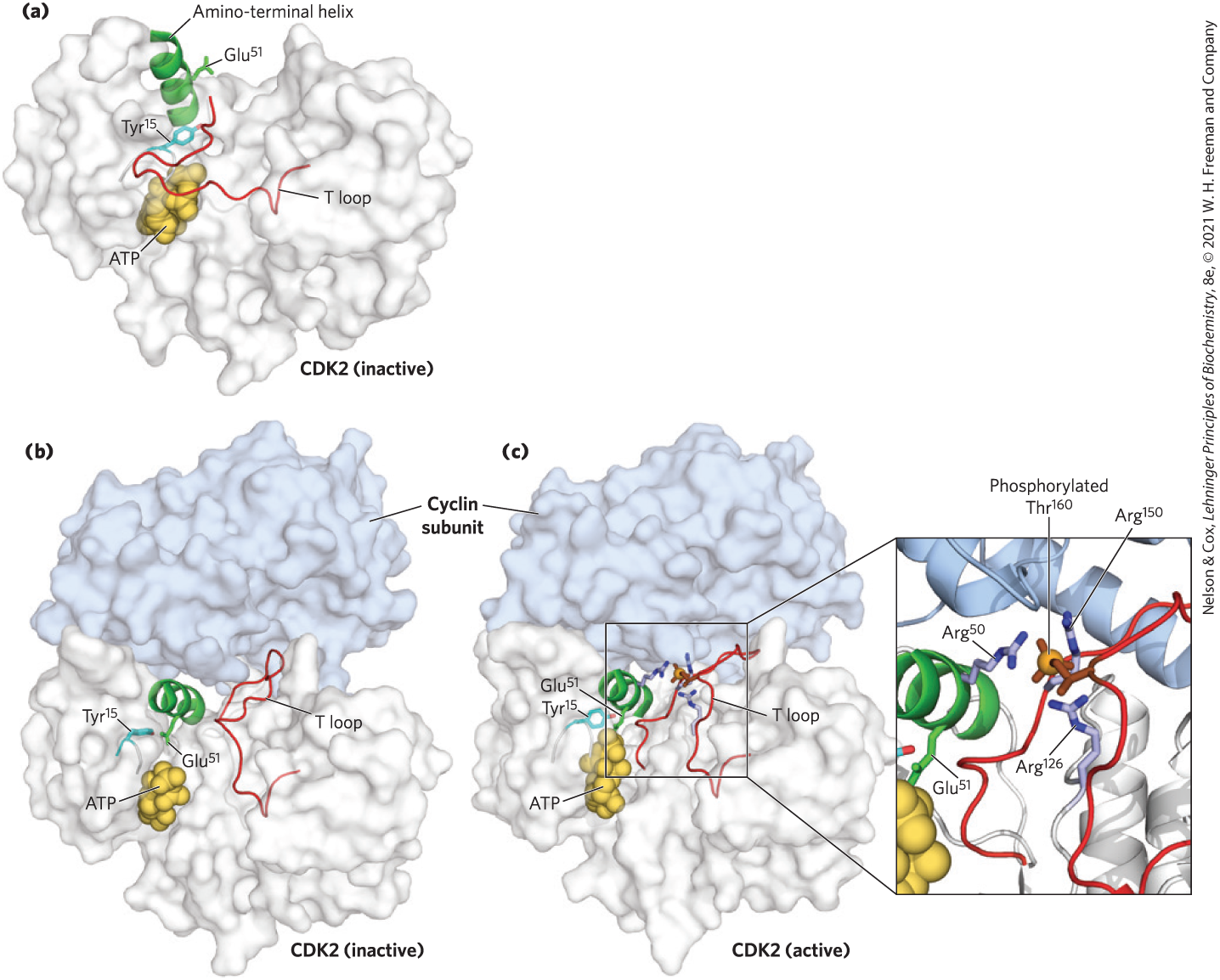
FIGURE 12-37 Activation of cyclin-dependent protein kinases (CDKs) by cyclin and phosphorylation. CDKs are active only when associated with a cyclin. The crystal structure of CDK2 with and without a cyclin reveals the basis for this activation. (a) Without the cyclin, CDK2 folds so that one segment, the T loop, obstructs the binding site for protein substrates. The binding site for ATP is also near the T loop and is blocked when is phosphorylated (not shown). (b) When the cyclin binds, it forces conformational changes that move the T loop away from the active site and reorient an amino-terminal helix, bringing a residue critical to catalysis into the active site. (c) When a Thr residue in the T loop is phosphorylated, its negative charges are stabilized by interaction with three Arg residues, holding the T loop away from the substrate-binding site. Removal of the phosphoryl group on gives ATP access to its binding site, fully activating CDK2 (see Fig. 12-38). [Data from (a) PDB ID 1HCK, U. Schulze-Gahmen et al., J. Med. Chem. 39:4540, 1996; (b) PDB ID 1FIN, P. D. Jeffrey et al., Nature 376:313, 1995; (c) PDB ID 1JST, A. A. Russo et al., Nature Struct. Biol. 3:696, 1996.]
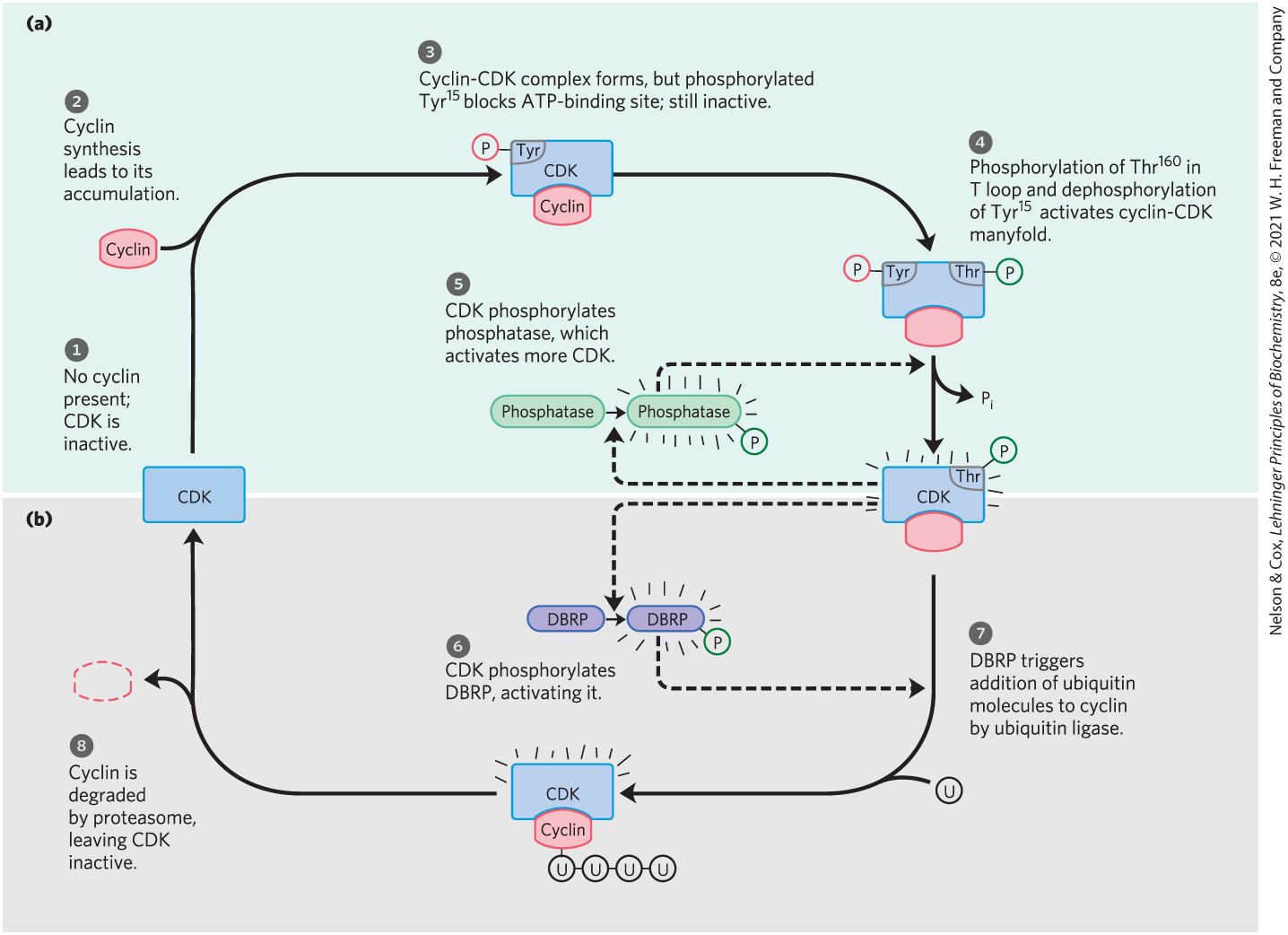
FIGURE 12-38 Regulation of CDK by phosphorylation and proteolysis. (a) The series of events that leads to activation of a cyclin-dependent protein kinase. (b) The periodic proteolytic degradation of cyclin, inactivating the cyclin-dependent protein kinase. The steps are described in the text.
The presence of a single-strand break in DNA signals arrest of the cell cycle in G2 by activating two proteins (ATM and ATR; see Fig. 12-40). These proteins trigger a cascade of responses that include inactivation of the PTP that dephosphorylates of the CDK. With the CDK inactivated, the cell is arrested in G2, unable to divide until the DNA is repaired and the effects of the cascade are reversed.
Highly specific and precisely timed proteolytic breakdown of mitotic cyclins regulates CDK activity throughout the cell cycle (Fig. 12-38b). How is the timing of cyclin breakdown controlled? A feedback loop occurs in the overall process shown in Figure 12-38. As a cell enters mitosis, the M-phase CDK is inactive (step ). As cyclin is synthesized (step ), the cyclin-CDK complex forms (step ). The T loop lies in the substrate-binding site of CDK, and - blocks its ATP-binding site, keeping the complex inactive. When in the T loop is phosphorylated, the loop moves out of the substrate-binding site, and when is dephosphorylated, ATP can bind. These two changes make the cyclin-CDK complex many times more active (step ). Further activation is achieved as CDK also phosphorylates and activates the enzyme that dephosphorylates - (step ). The active cyclin-CDK complex triggers its own inactivation by phosphorylation of DBRP (destruction box recognizing protein; step ). DBRP and ubiquitin ligase then attach several molecules of ubiquitin (U) to the cyclin (step ), targeting it for destruction by proteolytic enzyme complexes called proteasomes (step ). The role of ubiquitin and proteasomes is not limited to the regulation of cyclins; as we shall see in Chapter 27, both also take part in the turnover of cellular proteins, a process fundamental to cellular housekeeping.
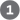 ). As cyclin is synthesized (step
). As cyclin is synthesized (step  ), the cyclin-CDK complex forms (step
), the cyclin-CDK complex forms (step  ). The T loop lies in the substrate-binding site of CDK, and
). The T loop lies in the substrate-binding site of CDK, and  ). Further activation is achieved as CDK also phosphorylates and activates the enzyme that dephosphorylates
). Further activation is achieved as CDK also phosphorylates and activates the enzyme that dephosphorylates 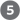 ). The active cyclin-CDK complex triggers its own inactivation by phosphorylation of DBRP (destruction box recognizing protein; step
). The active cyclin-CDK complex triggers its own inactivation by phosphorylation of DBRP (destruction box recognizing protein; step  ). DBRP and ubiquitin ligase then attach several molecules of
). DBRP and ubiquitin ligase then attach several molecules of  ), targeting it for destruction by proteolytic enzyme complexes called
), targeting it for destruction by proteolytic enzyme complexes called  ). The role of ubiquitin and proteasomes is not limited to the regulation of cyclins; as we shall see in
). The role of ubiquitin and proteasomes is not limited to the regulation of cyclins; as we shall see in The third mechanism for changing CDK activity is regulation of the rate of synthesis of the cyclin or CDK or both. Extracellular signals such as growth factors and cytokines (developmental signals that trigger cell division) activate, by phosphorylation, the nuclear transcription factors Jun and Fos, which promote the synthesis of many gene products, including cyclins, CDKs, and the transcription factor E2F. In turn, E2F stimulates production of several enzymes essential for the synthesis of deoxynucleotides and DNA, and the CDK and cyclin allow the cell to enter the S phase (Fig. 12-39).
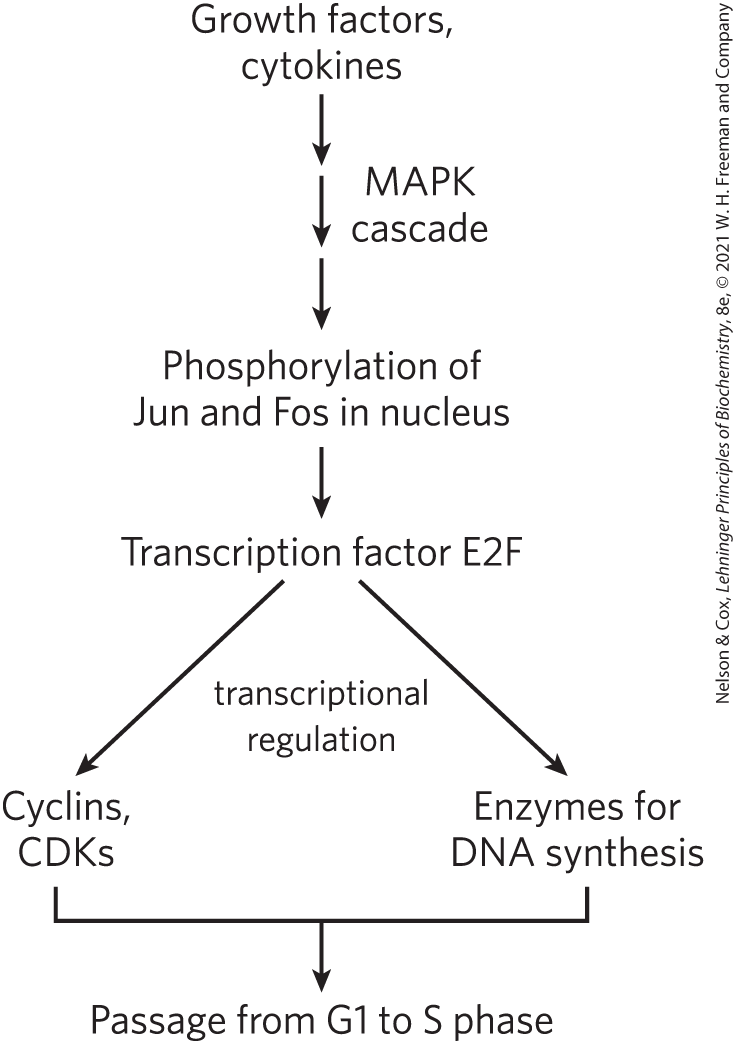
FIGURE 12-39 Regulation of cell division by growth factors.
Finally, specific protein inhibitors bind to and inactivate specific CDKs. One such protein is p21, which we discuss below.
These four control mechanisms modulate the activity of specific CDKs that, in turn, control whether a cell will divide, differentiate, become permanently quiescent, or begin a new cycle of division after a period of quiescence. The details of cell cycle regulation, such as the number of different cyclins and kinases and the combinations in which they act, differ from species to species, but the basic mechanism has been conserved in the evolution of all eukaryotic cells.
CDKs Regulate Cell Division by Phosphorylating Critical Proteins
We have examined how cells maintain close control of CDK activity, but how does the activity of CDKs control the cell cycle? There are scores of known CDK targets, and much remains to be learned. But we can see a general pattern behind CDK regulation by inspecting the effect of CDKs on the structures of lamin and myosin and on the activity of retinoblastoma protein.
The structure of the nuclear envelope is maintained in part by highly organized meshworks of intermediate filaments composed of the protein lamin. Breakdown of the nuclear envelope before segregation of the sister chromatids in mitosis is partly due to the phosphorylation of lamin by a CDK, which causes lamin filaments to depolymerize.
A second kinase target is the ATP-driven contractile machinery (actin and myosin) that pinches a dividing cell into two equal parts during cytokinesis. After the division, a CDK phosphorylates a small regulatory subunit of myosin, causing dissociation of myosin from actin filaments and inactivating the contractile machinery. Subsequent dephosphorylation allows reassembly of the contractile apparatus for the next round of cytokinesis.
A third and very important CDK substrate is the retinoblastoma protein, pRb; when DNA damage is detected, this protein participates in a mechanism that arrests cell division in G1 (Fig. 12-40). Named for the retinal tumor cell line in which it was discovered, pRb functions in most, perhaps all, cell types to regulate cell division in response to a variety of stimuli. Unphosphorylated pRb binds the transcription factor E2F; while bound to pRb, E2F cannot promote transcription of a group of genes necessary for DNA synthesis (the genes for DNA polymerase α, ribonucleotide reductase, and other proteins; see Chapter 25). In this state, the cell cycle cannot proceed from the G1 phase to the S phase, the step that commits a cell to mitosis and cell division. The pRb-E2F blocking mechanism is relieved when pRb is phosphorylated by cyclin E–CDK2, which occurs in response to a signal for cell division to proceed.
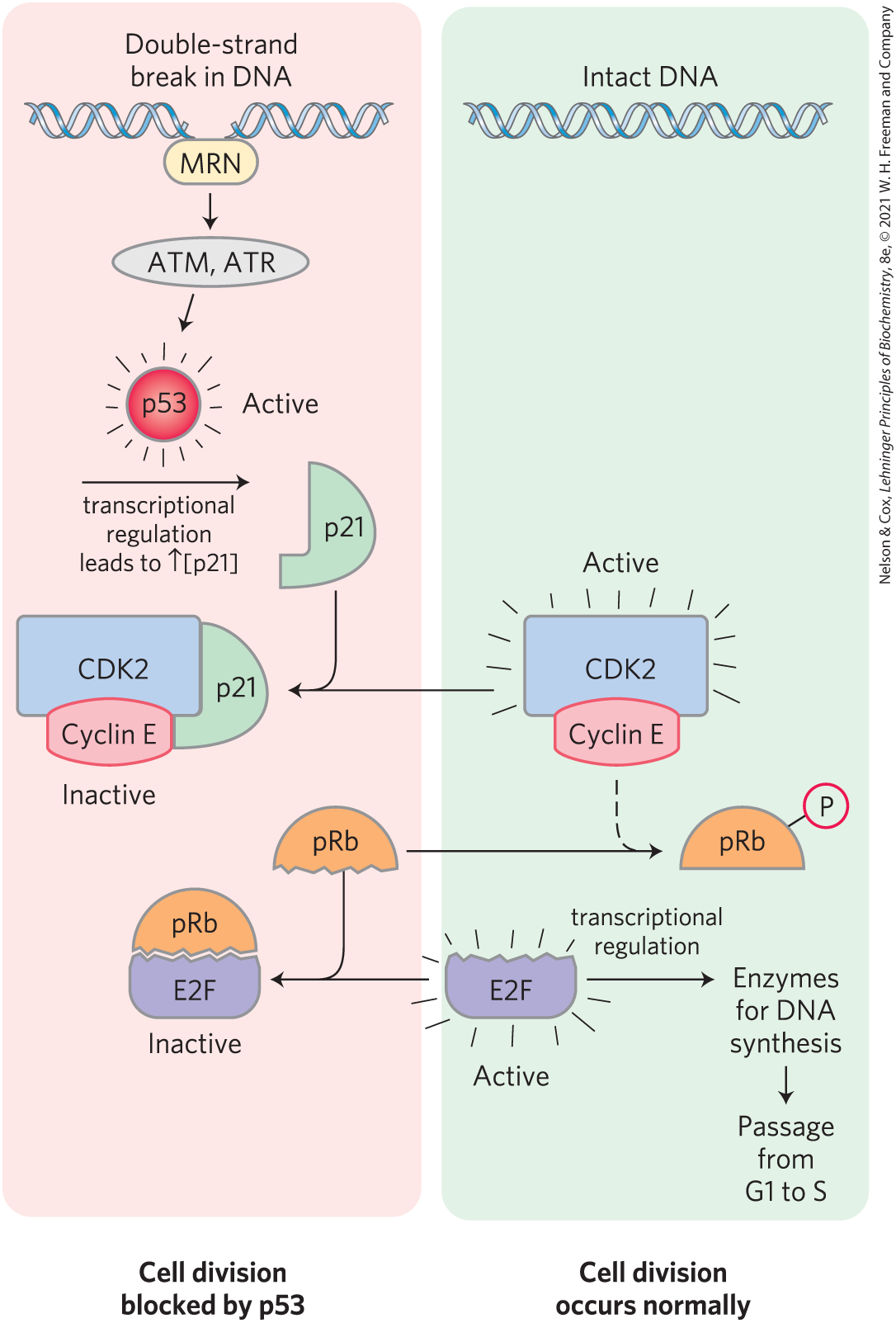
FIGURE 12-40 Regulation of passage from G1 to S by phosphorylation of pRb. Transcription factor E2F promotes transcription of genes for certain enzymes essential to DNA synthesis. The retinoblastoma protein, pRb, can bind E2F (lower left), inactivating it and preventing transcription of these genes. Phosphorylation of pRb by CDK2 prevents it from binding and inactivating E2F, and the genes are transcribed, allowing cell division. Damage to the cell’s DNA (upper left) triggers a series of events that inactivate CDK2, blocking cell division. When the protein MRN detects damage to the DNA, it activates two protein kinases, ATM and ATR, and they phosphorylate and activate the transcription factor p53. Active p53 promotes the synthesis of another protein, p21, an inhibitor of CDK2. Inhibition of CDK2 stops the phosphorylation of pRb, which therefore continues to bind and inhibit E2F. With E2F inactivated, genes essential to cell division are not transcribed and cell division is blocked. When DNA has been repaired, this inhibition is released, and the cell divides.
When the protein kinases ATM and ATR detect damage to DNA (signaled by the presence of the protein MRN at a double-strand break site), they phosphorylate p53, activating it to serve as a transcription factor that stimulates the synthesis of the protein p21 (Fig. 12-40). This protein inhibits the protein kinase activity of cyclin E–CDK2. In the presence of p21, pRb remains unphosphorylated and bound to E2F, blocking the activity of this transcription factor, and the cell cycle is arrested in G1. This gives the cell time to repair its DNA before entering the S phase, thereby avoiding the potentially disastrous transfer of a defective genome to one or both daughter cells. When the damage is too severe to allow effective repair, this same machinery triggers apoptosis (described below), a process that leads to the death of the cell, preventing the possible development of a cancer.
SUMMARY 12.8 Regulation of the Cell Cycle by Protein Kinases
- Progression through the cell cycle is regulated by the cyclin-dependent protein kinases (CDKs), which act at specific points in the cycle, phosphorylating key proteins and modulating their activities. The catalytic subunit of CDKs is inactive unless associated with the regulatory cyclin subunit.
- The activity of a cyclin-CDK complex changes during the cell cycle through differential synthesis of CDKs, specific degradation of the cyclin, phosphorylation and dephosphorylation of critical residues in CDKs, and binding of inhibitory proteins to specific cyclin-CDKs.
- A cyclin sequence (the destruction box) marks cyclin for tagging with ubiquitin and degradation in proteasomes. The rise of cyclin concentration by its synthesis ultimately triggers its degradation, yielding oscillations in cyclin level keyed to the cell cycle.
- Cells receive extracellular signals that determine the timing of their division. Scores of proteins are known targets of CDKs, many with unknown functions. Among the targets phosphorylated by cyclin-CDKs are proteins of the nuclear envelope and proteins required for cytokinesis and DNA repair.
 Progression through the cell cycle is regulated by the cyclin-dependent protein kinases (CDKs), which act at specific points in the cycle, phosphorylating key proteins and modulating their activities. The catalytic subunit of CDKs is inactive unless associated with the regulatory cyclin subunit.
Progression through the cell cycle is regulated by the cyclin-dependent protein kinases (CDKs), which act at specific points in the cycle, phosphorylating key proteins and modulating their activities. The catalytic subunit of CDKs is inactive unless associated with the regulatory cyclin subunit.