Most cells have only one or two sets of genomic DNA. Damaged proteins and RNA molecules can be quickly replaced by using information encoded in the DNA, but DNA molecules themselves are irreplaceable. Maintaining the integrity of the information in DNA is a cellular imperative, supported by an elaborate set of DNA repair systems. DNA can become damaged by a variety of processes, some spontaneous, others catalyzed by environmental agents (Chapter 8). Replication itself can very occasionally damage the information content in DNA when polymerase errors create mismatched base pairs (such as G paired with T).
The chemistry of DNA damage is diverse and complex. The cellular response to this damage includes enzymatic systems that catalyze some of the most interesting chemical transformations in DNA metabolism. We first examine the effects of alterations in DNA sequence and then consider specific repair systems.
Mutations Are Linked to Cancer
The best way to illustrate the importance of DNA repair is to consider the effects of unrepaired DNA damage (a lesion). The most serious outcome is a change in the base sequence of the DNA, which, if replicated and transmitted to future generations of cells, becomes permanent. A permanent change in the nucleotide sequence of DNA is called a mutation. Mutations can involve the replacement of one base pair with another (substitution mutation) or the addition or deletion of one or more base pairs (insertion or deletion mutations). If the mutation affects nonessential DNA or if it has a negligible effect on the function of a gene, it is known as a silent mutation. Rarely, a mutation confers some biological advantage. Most nonsilent mutations, however, are neutral or deleterious.
In mammals there is a strong correlation between the accumulation of mutations and cancer. A simple test developed by Bruce Ames in the 1970s measures the potential of a given chemical compound to promote certain easily detected mutations in a specialized bacterial strain (Fig. 25-19). Few of the chemicals that we encounter in daily life score as mutagens in this test. However, of the compounds known to be carcinogenic from extensive animal trials, more than 90% are also found to be mutagenic in the Ames test. Because of this strong correlation between mutagenesis and carcinogenesis, the Ames test for bacterial mutagens is still widely used as a rapid and inexpensive screen for potential human carcinogens.
FIGURE 25-19 Ames test for carcinogens, based on their mutagenicity. A strain of Salmonella typhimurium having a mutation that inactivates an enzyme of the histidine biosynthetic pathway is plated on a histidine-free medium. Few cells grow. (a) The few small colonies of S. typhimurium that do grow on a histidine-free medium carry spontaneous mutations that permit the histidine biosynthetic pathway to operate. Three identical nutrient plates (b), (c), and (d) have been inoculated with an equal number of cells. Each plate then receives a disk of filter paper containing progressively lower concentrations of a mutagen. The mutagen greatly increases the rate of back-mutation and hence the number of colonies. The clear areas around the filter paper indicate where the concentration of mutagen is so high that it is lethal to the cells. As the mutagen diffuses away from the filter paper, it is diluted to sublethal concentrations that promote back-mutation. Mutagens can be compared on the basis of their effect on mutation rate. Because many compounds undergo a variety of chemical transformations after entering cells, compounds are sometimes tested for mutagenicity after first incubating them with a liver extract. Some substances have been found to be mutagenic only after this treatment. [Bruce N. Ames, University of California, Berkeley, Department of Biochemistry and Molecular Biology.]
The genomic DNA in a typical mammalian cell accumulates many thousands of lesions during a 24-hour period. However, as a result of DNA repair, fewer than 1 in 1,000 become a mutation. DNA is a relatively stable molecule, but in the absence of repair systems, the cumulative effect of many infrequent but damaging reactions would make life impossible.
All Cells Have Multiple DNA Repair Systems
The number and diversity of repair systems reflect both the importance of DNA repair to cell survival and the diverse sources of DNA damage (Table 25-5). Some common types of lesions, such as pyrimidine dimers (see Fig. 8-30), can be repaired by several distinct systems. Nearly 200 genes in the human genome encode proteins dedicated to DNA repair. In many cases, the loss of function of one of these proteins results in genomic instability and an increased occurrence of oncogenesis (Box 25-1).
TABLE 25-5 Types of DNA Repair Systems in E. coli
Many DNA repair processes also seem to be extraordinarily inefficient energetically — an exception to the pattern observed in the vast majority of metabolic pathways, where every ATP is generally accounted for and used optimally. When the integrity of the genetic information is at stake, the amount of chemical energy invested in a repair process seems almost irrelevant.
Accurate DNA repair is possible largely because the DNA molecule consists of two complementary strands. Damaged DNA in one strand can be removed and replaced, without introducing mutations, by using the undamaged complementary strand as a template. We consider here the principal types of repair systems, beginning with those that repair the rare nucleotide mismatches that are left behind by replication.
Mismatch Repair
Correction of the rare mismatches left after replication in E. coli improves the overall fidelity of replication by an additional factor of . The mismatches are nearly always corrected to reflect the information in the old (template) strand, which the repair system can distinguish from the newly synthesized strand by the presence of methyl group tags on the template DNA. The mismatch repair system of E. coli includes at least 10 protein components (Table 25-5) that function either in strand discrimination or in the repair process itself. The functions of many of these were first worked out by Paul Modrich and colleagues in the 1980s.
The strand discrimination mechanism has not been determined for most bacteria or eukaryotes, but it is well understood for E. coli and some closely related bacterial species. In these bacteria, strand discrimination is based on the action of Dam methylase, which, as you will recall, methylates DNA at the position of all adenines within GATC sequences. Immediately after passage of the replication fork, there is a short period (a few seconds or minutes) during which the template strand is methylated but the newly synthesized strand is not (Fig. 25-20). The transient unmethylated state of GATC sequences in the newly synthesized strand permits the new strand to be distinguished from the template strand. Replication mismatches in the vicinity of a hemimethylated GATC sequence are then repaired according to the information in the methylated parent (template) strand. If both strands are methylated at a GATC sequence, few mismatches are repaired; if neither strand is methylated, repair occurs but does not favor either strand. The methyl-directed mismatch repair system of E. coli efficiently repairs mismatches up to 1,000 bp from a hemimethylated GATC sequence.
FIGURE 25-20 Methylation and mismatch repair. Methylation of DNA strands can serve to distinguish parent (template) strands from newly synthesized strands in E. coli DNA, a function that is critical to mismatch repair. The methylation occurs at the of adenines in sequences. This sequence is a palindrome, present in opposite orientations on the two strands.
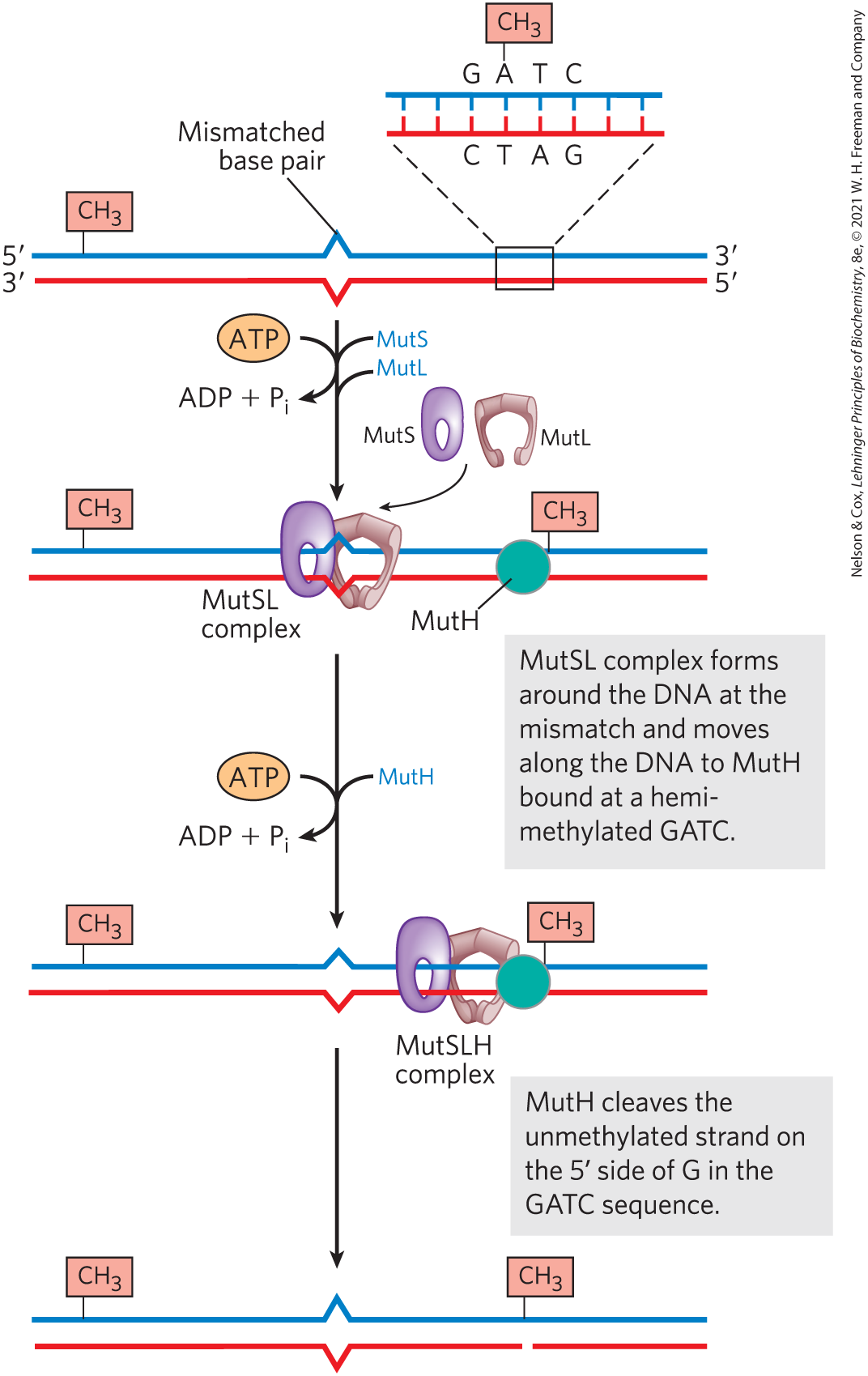
How is the mismatch correction process directed by relatively distant GATC sequences? Figure 25-21 illustrates one mechanism. MutS scans the DNA and forms a clamplike complex upon encountering a lesion. The complex binds to all mismatched base pairs (except C–C). MutL protein forms a complex with MutS protein, and the MutSL complex slides along the DNA to find a hemimethylated GATC sequence. MutH binds to MutL, and the MutSLH complex moves in either direction at random along the DNA. MutH has a site-specific endonuclease activity that is inactive until the complex encounters a hemimethylated GATC sequence. At this site, MutH catalyzes cleavage of the unmethylated strand on the side of the G in GATC, which marks the strand for repair. Further steps in the pathway depend on where the mismatch is located relative to this cleavage site (Fig. 25-22).
FIGURE 25-21 A model for the early steps of methyl-directed mismatch repair. Recognition of the sequence and of the mismatch are specialized functions of the MutH and MutS proteins, respectively.
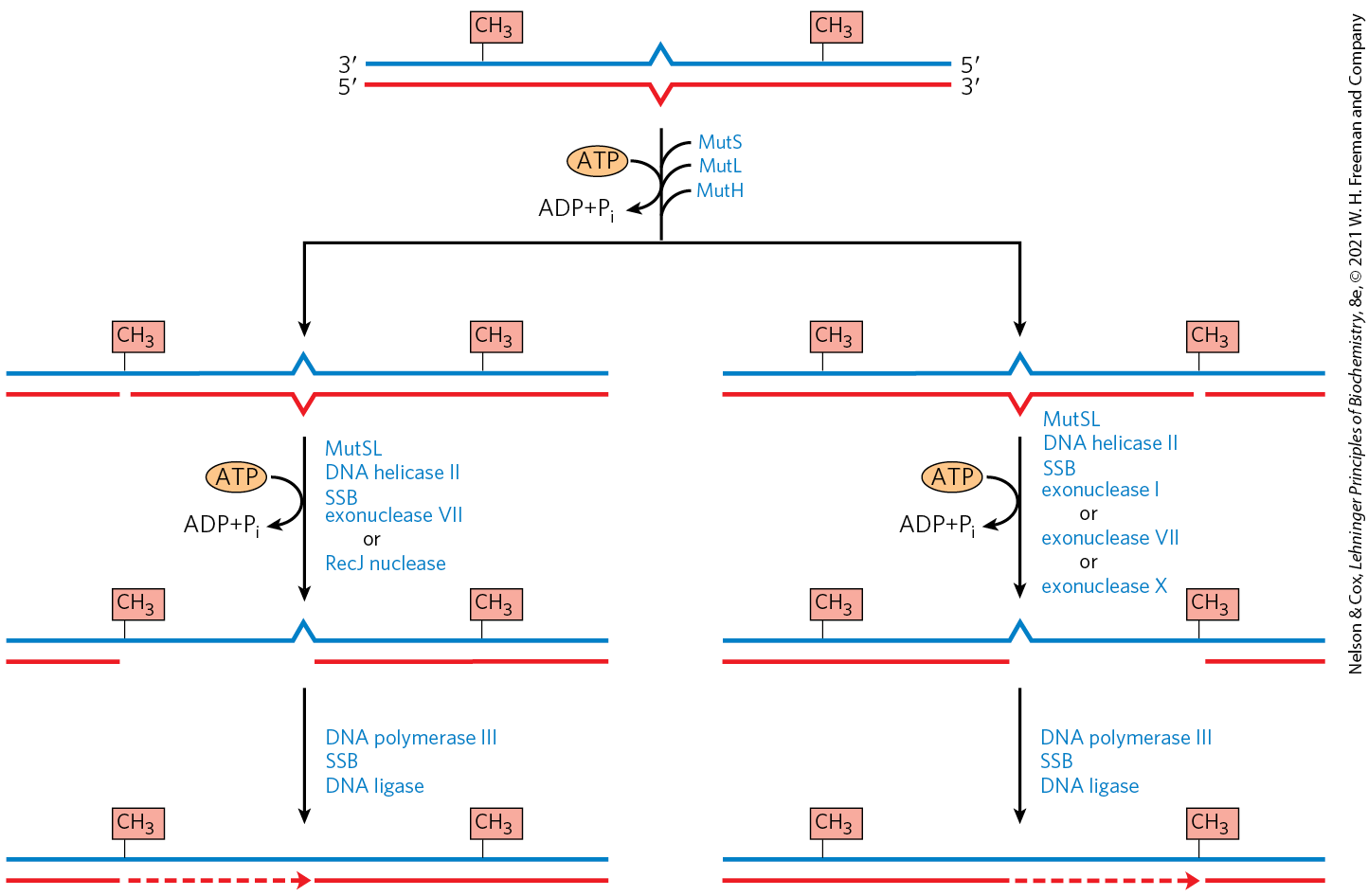
FIGURE 25-22 Completion of methyl-directed mismatch repair. The combined action of DNA helicase II, SSB, and one of four different exonucleases removes a segment of the new strand between the MutH cleavage site and a point just beyond the mismatch. The particular exonuclease depends on the location of the cleavage site relative to the mismatch, as shown by the alternative pathways here. The resulting gap is filled in (dashed line) by DNA polymerase III, and the nick is sealed by DNA ligase (not shown).
When the mismatch is on the side of the cleavage site (Fig. 25-22, right side), the unmethylated strand is unwound and degraded in the direction from the cleavage site through the mismatch, and this segment is replaced with new DNA. This process requires the combined action of DNA helicase II (also called UvrD helicase), SSB, exonuclease I or exonuclease X (both of which degrade strands of DNA in the direction) or exonuclease VII (which degrades single-stranded DNA in either direction), DNA polymerase III, and DNA ligase. The pathway for repair of mismatches on the side of the cleavage site is similar (Fig. 25-22, left), except that the exonuclease is either exonuclease VII or RecJ nuclease (which degrades single-stranded DNA in the direction).
Mismatch repair is particularly costly for E. coli in terms of energy expended. The mismatch may occur 1,000 or more base pairs from the GATC sequence. The degradation and replacement of a strand segment of this length require an enormous investment in activated deoxynucleotide precursors to repair a single mismatched base. This again underscores the importance to the cell of genomic integrity.
Eukaryotic cells also have mismatch repair systems, with several proteins structurally and functionally analogous to the bacterial MutS and MutL (but not MutH) proteins. Alterations in human genes encoding proteins of this type produce some of the most common inherited cancer-susceptibility syndromes (see Box 25-1), further demonstrating the value to the organism of DNA repair systems. The main MutS homologs in most eukaryotes, from yeast to humans, are MSH2 (MutS homolog), MSH3, and MSH6. Heterodimers of MSH2 and MSH6 generally bind to single base-pair mismatches, and they bind less well to slightly longer mispaired loops. In many organisms, the longer mismatches (2 to 6 bp) may be bound instead by a heterodimer of MSH2 and MSH3, or are bound by both types of heterodimers in tandem. Homologs of MutL, predominantly a heterodimer of MLH1 (MutL homolog) and PMS1 (post-meiotic segregation), bind to and stabilize the MSH complexes. Many details of the subsequent events in eukaryotic mismatch repair remain to be worked out. In particular, we do not know how newly synthesized DNA strands are identified, although research reveals that this process does not involve GATC sequences.
Base-Excision Repair
Every cell has a class of enzymes called DNA glycosylases that recognize particularly common DNA lesions (such as the products of cytosine and adenine deamination; see Fig. 8-29a) and remove the affected base by cleaving the N-glycosyl bond. The repair pathway is called base-excision repair, as the first step involves only the removal of the base rather than an entire nucleotide. The cleavage creates an apurinic or apyrimidinic site in the DNA, commonly referred to as an AP site or abasic site. Each DNA glycosylase is generally specific for one type of lesion.
Uracil DNA glycosylases, for example, found in most cells, specifically remove from DNA the uracil that results from spontaneous deamination of cytosine. Mutant cells that lack this enzyme have a high rate of to mutations. This glycosylase does not remove uracil residues from RNA or thymine residues from DNA. The capacity to distinguish thymine from uracil, the product of cytosine deamination — necessary for the selective repair of the latter — may be one reason why DNA evolved to contain thymine instead of uracil (p. 280).
Most bacteria have just one type of uracil DNA glycosylase, whereas humans have at least four types, with different specificities — an indicator of the importance of removing uracil from DNA. The most abundant human uracil glycosylase, UNG, is associated with the replisome, where it eliminates the occasional U residue inserted in place of a T during replication. The deamination of C residues is 100-fold faster in single-stranded DNA than in double-stranded DNA, and humans have an enzyme, hSMUG1, that removes any U residues occurring in single-stranded DNA during replication or transcription. Two other human DNA glycosylases, TDG and MBD4, remove either U or T residues paired with G, which are generated by deamination of cytosine or 5-methylcytosine, respectively.
Other DNA glycosylases recognize and remove a variety of damaged bases, including formamidopyrimidine and 8-hydroxyguanine (both arising from purine oxidation), hypoxanthine (from adenine deamination), and alkylated bases such as 3-methyladenine and 7-methylguanine. Glycosylases that recognize other lesions, including pyrimidine dimers, have also been identified in some classes of organisms. Remember that AP sites also arise from slow, spontaneous hydrolysis of the N-glycosyl bonds in DNA (see Fig. 8-29b).
Once an AP site has been formed by a DNA glycosylase, another type of enzyme must repair it. The repair is not made by simply inserting a new base and re-forming the N-glycosyl bond. Instead, the deoxyribose -phosphate left behind is removed and replaced with a new nucleotide. This process begins with one of the AP endonucleases, enzymes that cut the DNA strand containing the AP site. The position of the incision relative to the AP site ( or to the site) depends on the type of AP endonuclease. A segment of DNA including the AP site is then removed, DNA polymerase I replaces the DNA, and DNA ligase seals the remaining nick (Fig. 25-23). In eukaryotes, nucleotide replacement is carried out by specialized polymerases, as described below.
FIGURE 25-23 DNA repair by the base-excision repair pathway. A DNA glycosylase recognizes a damaged base (in this case, a uracil) and cleaves between the base and deoxyribose in the backbone. An AP endonuclease cleaves the phosphodiester backbone near the AP site. DNA polymerase I initiates repair synthesis from the free hydroxyl at the nick, removing (with its exonuclease activity) and replacing a portion of the damaged strand. The nick remaining after DNA polymerase I has dissociated is sealed by DNA ligase.
Nucleotide-Excision Repair
DNA lesions that cause large distortions in the helical structure of DNA generally are repaired by the nucleotide-excision system, a repair pathway critical to the survival of all free-living organisms. In nucleotide-excision repair (Fig. 25-24), a multisubunit enzyme (excinuclease) hydrolyzes two phosphodiester bonds, one on either side of the distortion caused by the lesion. In E. coli and other bacteria, the enzyme system hydrolyzes the fifth phosphodiester bond on the side and the eighth phosphodiester bond on the side to generate a fragment of 12 to 13 nucleotides (depending on whether the lesion involves one or two bases). In humans and other eukaryotes, the enzyme system hydrolyzes the sixth phosphodiester bond on the side and the twenty-second phosphodiester bond on the side, producing a fragment of 27 to 29 nucleotides. Following the dual incision, the excised oligonucleotides are released from the duplex and the resulting gap is filled — by DNA polymerase I in E. coli and DNA polymerase ε in humans. DNA ligase seals the nick.
FIGURE 25-24 Nucleotide-excision repair in E. coli and humans. The general pathway of nucleotide-excision repair is similar in all organisms. An excinuclease binds to DNA at the site of a bulky lesion and cleaves the damaged DNA strand on either side of the lesion. The DNA segment — of 13 nucleotides (13mer) or 29 nucleotides (29mer) — is removed with the aid of a helicase. The gap is filled in by DNA polymerase, and the remaining nick is sealed with DNA ligase. [Information from a figure provided by Aziz Sancar.]
In E. coli, the key enzymatic complex is the ABC excinuclease, which has three protein components, UvrA , UvrB , and UvrC . The term “excinuclease” is used to describe the unique capacity of this enzyme complex to catalyze two specific endonucleolytic cleavages, distinguishing this activity from that of standard endonucleases. A dimeric UvrA protein (an ATPase) scans the DNA and binds to the site of a lesion. A UvrB protein can bind to UvrA either before or after an encounter with the lesion. At the lesion, the UvrA dimer dissociates, leaving a tight UvrB-DNA complex. UvrC protein then binds to UvrB, and UvrB makes an incision at the fifth phosphodiester bond on the side of the lesion. This is followed by a UvrC-mediated incision at the eighth phosphodiester bond on the side. The resulting fragment, consisting of 12 to 13 nucleotides, is removed by UvrD helicase. The short gap thus created is filled in by DNA polymerase I and DNA ligase. This pathway (Fig. 25-24, left) is a primary repair route for many types of lesions, including cyclobutane pyrimidine dimers, 6-4 photoproducts (see Fig. 8-30), and several other types of base adducts, including benzo pyrene-guanine, which is formed in DNA by exposure to cigarette smoke. The nucleolytic activity of the ABC excinuclease is novel in the sense that two cuts are made in the DNA.
The mechanism of eukaryotic excinucleases is quite similar to that of the bacterial enzyme, although at least 16 polypeptides with no similarity to the E. coli excinuclease subunits are required for the dual incision. Some of the nucleotide-excision repair and base-excision repair in eukaryotes is closely tied to transcription (see Chapter 26). Genetic deficiencies in nucleotide-excision repair in humans give rise to a variety of serious diseases (see Box 25-1).
Direct Repair
Several types of damage are repaired without removing a base or nucleotide. The best-characterized example is direct photoreactivation of cyclobutane pyrimidine dimers, a reaction promoted by DNA photolyases. Pyrimidine dimers result from a UV-induced reaction. Through a mechanism worked out by Aziz Sancar and colleagues, photolyases use energy derived from absorbed light to reverse the damage (Fig. 25-25). Photolyases generally contain two cofactors that serve as light-absorbing agents, or chromophores: in all organisms, one is ; in E. coli and yeast, the other is a folate. The reaction mechanism entails the generation of free radicals. DNA photolyases are not found in humans and other placental mammals.
MECHANISM FIGURE 25-25 Repair of pyrimidine dimers with photolyase. Energy derived from absorbed light is used to reverse the photoreaction that caused the lesion. The two chromophores in E. coli photolyase , (MTHFpolyGlu) and , perform complementary functions. MTHFpolyGlu functions as a photoantenna to absorb blue-light photons. The excitation energy passes to , and the excited flavin donates an electron to the pyrimidine dimer, leading to the rearrangement as shown.
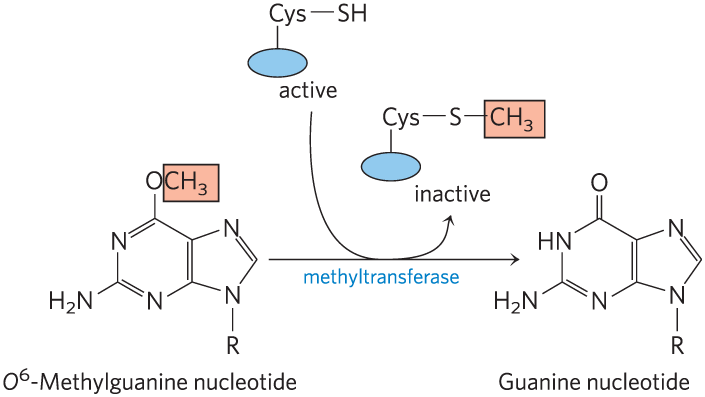
Additional examples are seen in the repair of nucleotides with alkylation damage. The modified nucleotide -methylguanine forms in the presence of alkylating agents and is a common and highly mutagenic lesion. It tends to pair with thymine rather than cytosine during replication, and therefore causes to mutations (Fig. 25-26). Direct repair of -methylguanine is carried out by -methylguanine-DNA methyltransferase, a protein that catalyzes transfer of the methyl group of -methylguanine to one of its own Cys residues. This methyltransferase is not strictly an enzyme, because a single methyl transfer event permanently methylates the protein, inactivating it in this pathway. The consumption of an entire protein molecule to correct a single damaged base is another vivid illustration of the priority given to maintaining the integrity of cellular DNA.
FIGURE 25-26 Example of how DNA damage results in mutations. (a) The methylation product -methylguanine pairs with thymine rather than cytosine residues. (b) If not repaired, this leads to a to mutation after replication.
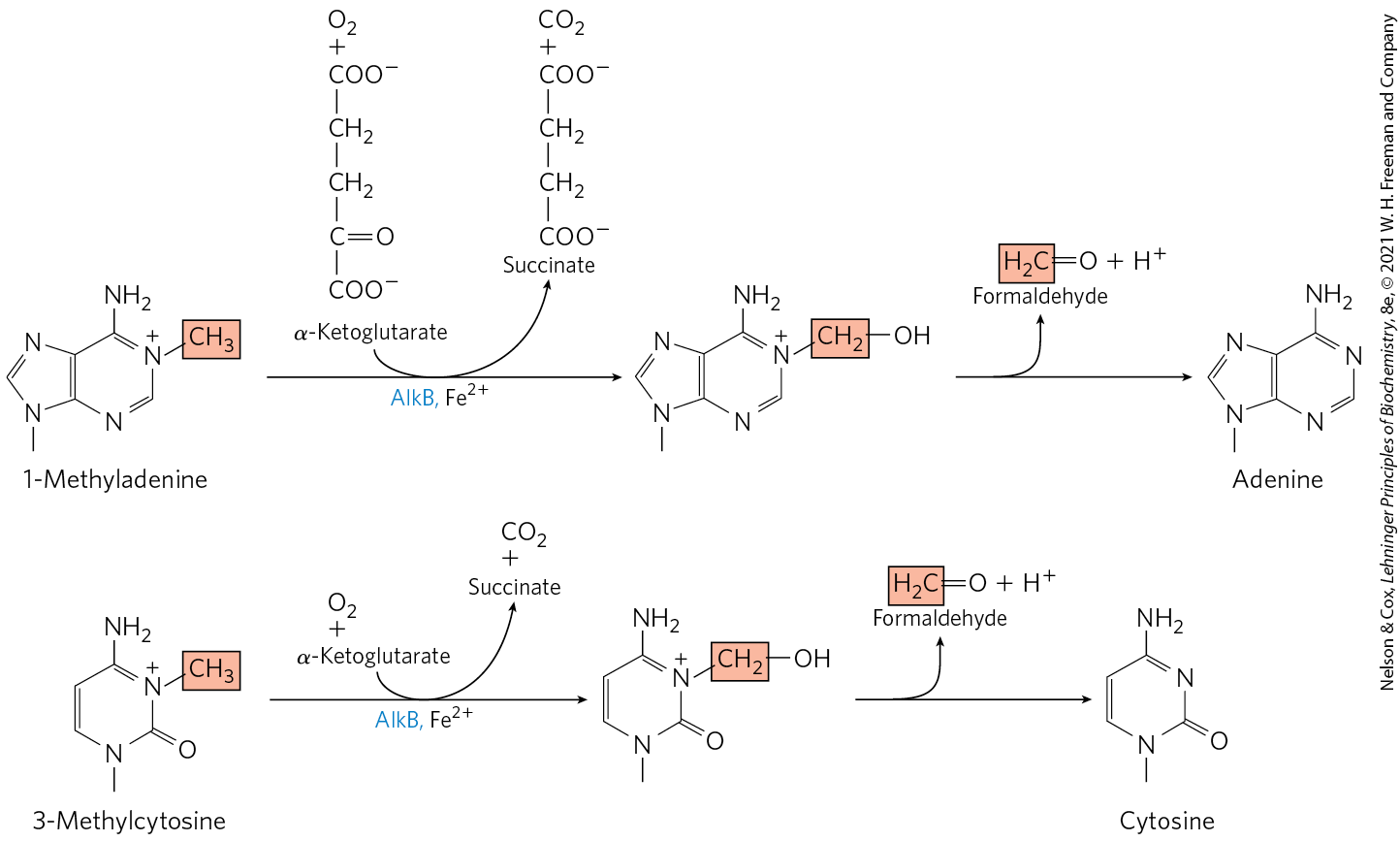
A very different but equally direct mechanism is used to repair 1-methyladenine and 3-methylcytosine. The amino groups of A and C residues are sometimes methylated when the DNA is single-stranded, and the methylation directly affects proper base pairing. In E. coli, oxidative demethylation of these alkylated nucleotides is mediated by the AlkB protein, a member of the dioxygenase superfamily (Fig. 25-27). (See Box 4-2 for a description of proline hydroxylation, catalyzed by another member of this enzyme family.)
FIGURE 25-27 Direct repair of alkylated bases by AlkB. The AlkB protein is an α-ketoglutarate-–dependent hydroxylase (see Box 4-2). It catalyzes the oxidative demethylation of 1-methyladenine and 3-methylcytosine residues.
The Interaction of Replication Forks with DNA Damage Can Lead to Error-Prone Translesion DNA Synthesis
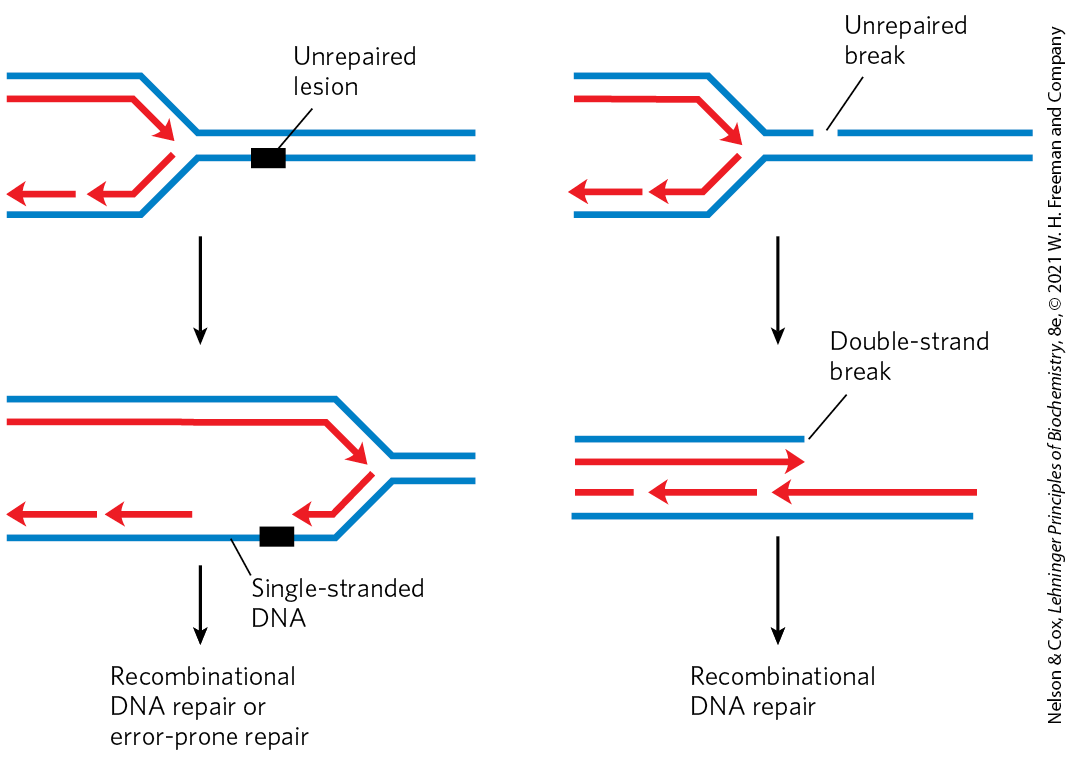
The repair pathways considered to this point generally work only for lesions in double-stranded DNA, the undamaged strand providing the correct genetic information to restore the damaged strand to its original state. However, in certain types of lesions, such as double-strand breaks, double-strand cross-links, or lesions in a single-stranded DNA, the complementary strand is itself damaged or is absent. Double-strand breaks and lesions in single-stranded DNA most often arise when a replication fork encounters an unrepaired DNA lesion (Fig. 25-28). Such lesions and DNA cross-links can also result from ionizing radiation and oxidative reactions.
FIGURE 25-28 DNA damage and its effect on DNA replication. If the replication fork encounters an unrepaired lesion or strand break, the DNA polymerase sometimes disengages and re-initiates downstream. The lesion remains in an unreplicated, single-stranded gap that is left behind the replication fork (left). In other cases, a replication fork may encounter a lesion that is actively undergoing repair such that a transient break is present in one of the template strands. When the replication fork encounters it, the single-strand break becomes a double-strand break (right). In each case, the damage to one strand cannot be repaired by mechanisms described earlier in this chapter, because the complementary strand required to direct accurate repair is damaged or absent. There are at least two possible avenues for repair: recombinational DNA repair or, when lesions are unusually numerous, error-prone repair. The latter mechanism involves translesion DNA polymerases such as DNA polymerase V, encoded by the umuC and umuD genes and activated by the RecA protein that can replicate, albeit inaccurately, over many types of lesions. The repair mechanism is “error-prone” because mutations often result.
At a stalled bacterial replication fork, there are two avenues for repair. In the absence of a second strand, the information required for accurate repair must come from a separate, homologous chromosome. The repair system thus involves homologous genetic recombination. This recombinational DNA repair is considered in detail in Section 25.3. Under some conditions, a second repair pathway, error-prone translesion DNA synthesis (often abbreviated TLS), becomes available. When this pathway is active, DNA repair is significantly less accurate, and a high mutation rate can result. In bacteria, error-prone translesion DNA synthesis is part of a cellular stress response to extensive DNA damage known, appropriately enough, as the SOS response. Some of the 40 or more SOS proteins, such as the UvrA and UvrB proteins involved in the error-free nucleotide-excision repair already described, are normally present in the cell but are induced to higher levels as part of the SOS response. Additional SOS proteins participate in the pathway for error-prone repair; these include the UmuC and UmuD proteins (unmutable; lack of the umu gene eliminates error-prone repair). The UmuD protein is cleaved in an SOS-regulated process to a shorter form called , which forms a complex with UmuC and a protein called RecA (described in Section 25.3) to create a specialized DNA polymerase, DNA polymerase V , which can replicate past many of the DNA lesions that would normally block replication. Proper base pairing is often impossible at the site of such a lesion, so this translesion replication is error-prone.
Given the emphasis on the importance of genomic integrity throughout this chapter, the existence of a system that increases the rate of mutation may seem incongruous. However, we can think of this system as a desperation strategy. The umuC and umuD genes are fully induced only late in the SOS response, and they are not activated for translesion synthesis initiated by UmuD cleavage unless the levels of DNA damage are particularly high and all replication forks are blocked. The mutations resulting from DNA polymerase V–mediated replication kill some cells and create deleterious mutations in others, but this is the biological price a species pays to overcome an otherwise insurmountable barrier to replication, as it permits at least a few mutant daughter cells to survive. The resultant mutations contribute to evolution.
Yet another DNA polymerase, DNA polymerase IV, is also induced during the SOS response. Replication by DNA polymerase IV, a product of the dinB gene, is also highly error-prone. The bacterial DNA polymerases IV and V (Table 25-1) are part of a family of TLS polymerases found in all organisms. These enzymes lack a proofreading exonuclease and have a more open active site than other DNA polymerases, one that accommodates damaged template nucleotides. With these enzymes, the fidelity of base selection during replication may be reduced by a factor of , lowering overall replication fidelity to one error in ∼1,000 nucleotides.
Mammals have many low-fidelity DNA polymerases of the TLS polymerase family. However, the presence of these enzymes does not necessarily translate into an unacceptable mutational burden, because most of the enzymes also have specialized functions in DNA repair. DNA polymerase (eta), for example, found in all eukaryotes, promotes translesion synthesis primarily across cyclobutane T–T dimers. Few mutations result, because the enzyme preferentially inserts two A residues across from the linked T residues. Several other low-fidelity polymerases, including DNA polymerases β, (iota), and , have specialized roles in eukaryotic base-excision repair. Each of these enzymes has a -deoxyribose phosphate lyase activity in addition to its polymerase activity. After base removal by a glycosylase and backbone cleavage by an AP endonuclease, these polymerases remove the abasic site (a -deoxyribose phosphate) and fill in the very short gap. The frequency of mutation due to DNA polymerase activity is minimized by the short lengths (often one nucleotide) of DNA synthesized.
What emerges from research into cellular DNA repair systems is a picture of a DNA metabolism that maintains genomic integrity with multiple and often redundant systems. Most of the major DNA repair systems occur in all organisms. These repair systems are often integrated with the DNA replication systems and are complemented by recombination systems, which we turn to next.
SUMMARY 25.2 DNA Repair
Mutations are genomic changes that alter the information in DNA. When mutations occur in genes encoding enzymes involved in DNA repair, the loss of function can lead to cancer.
Cells have many systems for DNA repair. Major repair systems present in all organisms include mismatch repair, base excision repair, nucleotide-excision repair, and direct repair.
In bacteria, TLS DNA polymerases respond to very heavy DNA damage with error-prone translesion DNA synthesis. In eukaryotes, similar polymerases have specialized roles in DNA repair that minimize the introduction of mutations.
 Damaged proteins and RNA molecules can be quickly replaced by using information encoded in the DNA, but DNA molecules themselves are irreplaceable. Maintaining the integrity of the information in DNA is a cellular imperative, supported by an elaborate set of DNA repair systems. DNA can become damaged by a variety of processes, some spontaneous, others catalyzed by environmental agents (
Damaged proteins and RNA molecules can be quickly replaced by using information encoded in the DNA, but DNA molecules themselves are irreplaceable. Maintaining the integrity of the information in DNA is a cellular imperative, supported by an elaborate set of DNA repair systems. DNA can become damaged by a variety of processes, some spontaneous, others catalyzed by environmental agents ( The best way to illustrate the importance of DNA repair is to consider the effects of unrepaired DNA damage (a lesion).
The best way to illustrate the importance of DNA repair is to consider the effects of unrepaired DNA damage (a lesion).  The most serious outcome is a change in the base sequence of the DNA, which, if replicated and transmitted to future generations of cells, becomes permanent. A permanent change in the nucleotide sequence of DNA is called a mutation. Mutations can involve the replacement of one base pair with another (substitution mutation) or the addition or deletion of one or more base pairs (insertion or deletion mutations). If the mutation affects nonessential DNA or if it has a negligible effect on the function of a gene, it is known as a
The most serious outcome is a change in the base sequence of the DNA, which, if replicated and transmitted to future generations of cells, becomes permanent. A permanent change in the nucleotide sequence of DNA is called a mutation. Mutations can involve the replacement of one base pair with another (substitution mutation) or the addition or deletion of one or more base pairs (insertion or deletion mutations). If the mutation affects nonessential DNA or if it has a negligible effect on the function of a gene, it is known as a 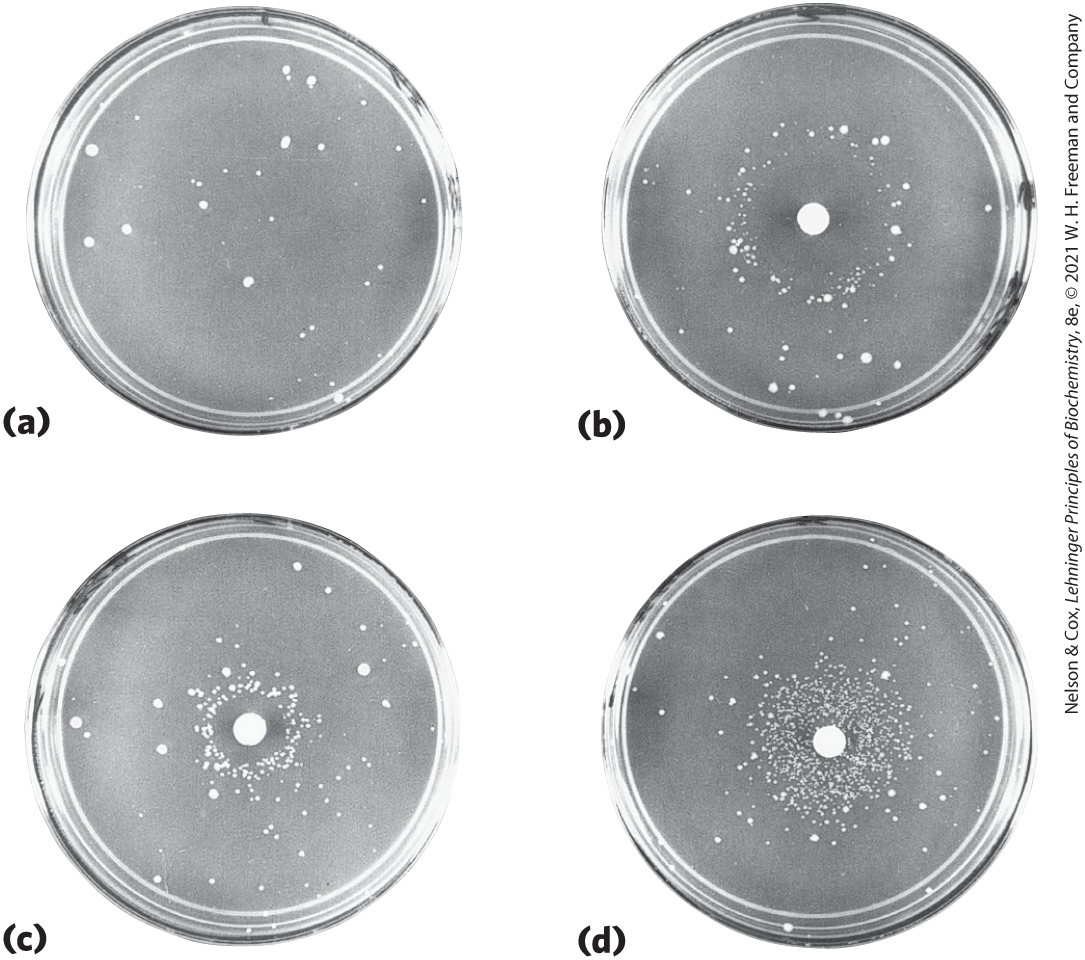


 Many DNA repair processes also seem to be extraordinarily inefficient energetically — an exception to the pattern observed in the vast majority of metabolic pathways, where every ATP is generally accounted for and used optimally. When the integrity of the genetic information is at stake, the amount of chemical energy invested in a repair process seems almost irrelevant.
Many DNA repair processes also seem to be extraordinarily inefficient energetically — an exception to the pattern observed in the vast majority of metabolic pathways, where every ATP is generally accounted for and used optimally. When the integrity of the genetic information is at stake, the amount of chemical energy invested in a repair process seems almost irrelevant.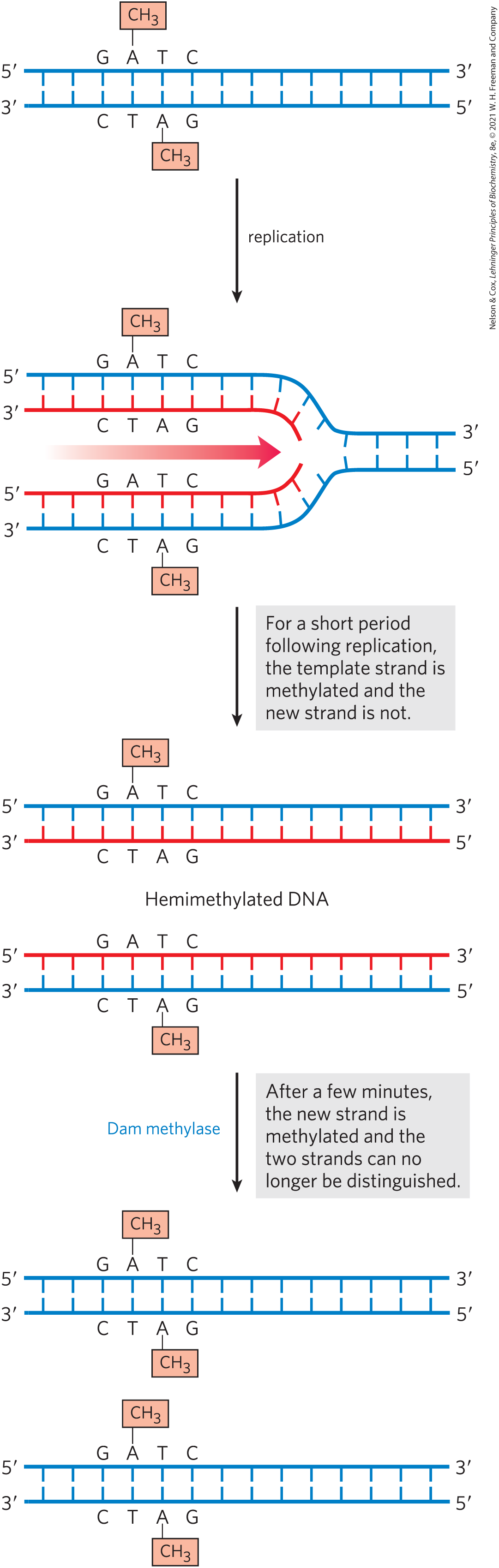
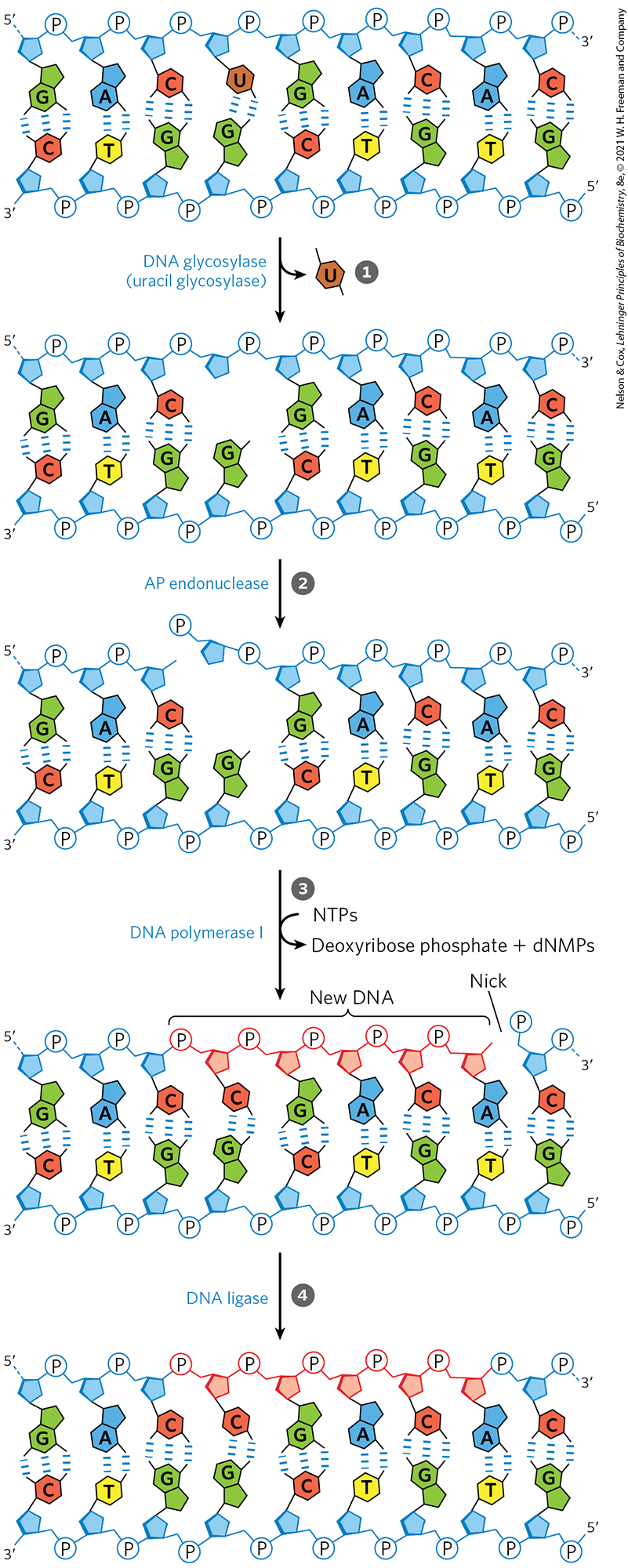
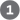 A DNA glycosylase recognizes a damaged base (in this case, a uracil) and cleaves between the base and deoxyribose in the backbone.
A DNA glycosylase recognizes a damaged base (in this case, a uracil) and cleaves between the base and deoxyribose in the backbone.  An AP endonuclease cleaves the phosphodiester backbone near the AP site.
An AP endonuclease cleaves the phosphodiester backbone near the AP site.  DNA polymerase I initiates repair synthesis from the free hydroxyl at the nick, removing (with its exonuclease activity) and replacing a portion of the damaged strand.
DNA polymerase I initiates repair synthesis from the free hydroxyl at the nick, removing (with its exonuclease activity) and replacing a portion of the damaged strand.  The nick remaining after DNA polymerase I has dissociated is sealed by DNA ligase.
The nick remaining after DNA polymerase I has dissociated is sealed by DNA ligase.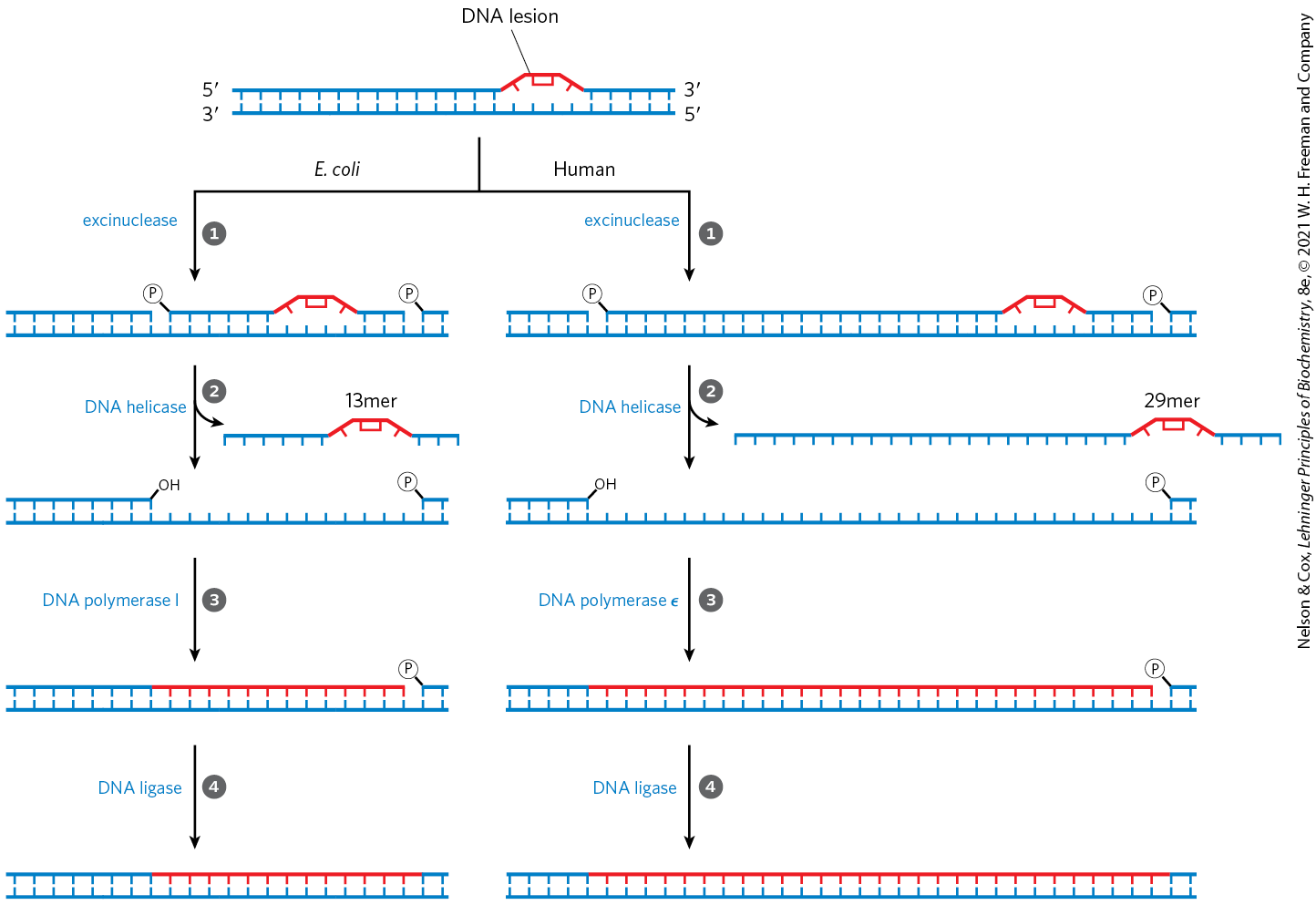

 Mammals have many low-fidelity DNA polymerases of the TLS polymerase family. However, the presence of these enzymes does not necessarily translate into an unacceptable mutational burden, because most of the enzymes also have specialized functions in DNA repair. DNA polymerase (eta), for example, found in all eukaryotes, promotes translesion synthesis primarily across cyclobutane T–T dimers. Few mutations result, because the enzyme preferentially inserts two A residues across from the linked T residues. Several other low-fidelity polymerases, including DNA polymerases β, (iota), and , have specialized roles in eukaryotic base-excision repair. Each of these enzymes has a -deoxyribose phosphate lyase activity in addition to its polymerase activity. After base removal by a glycosylase and backbone cleavage by an AP endonuclease, these polymerases remove the abasic site (a -deoxyribose phosphate) and fill in the very short gap. The frequency of mutation due to DNA polymerase activity is minimized by the short lengths (often one nucleotide) of DNA synthesized.
Mammals have many low-fidelity DNA polymerases of the TLS polymerase family. However, the presence of these enzymes does not necessarily translate into an unacceptable mutational burden, because most of the enzymes also have specialized functions in DNA repair. DNA polymerase (eta), for example, found in all eukaryotes, promotes translesion synthesis primarily across cyclobutane T–T dimers. Few mutations result, because the enzyme preferentially inserts two A residues across from the linked T residues. Several other low-fidelity polymerases, including DNA polymerases β, (iota), and , have specialized roles in eukaryotic base-excision repair. Each of these enzymes has a -deoxyribose phosphate lyase activity in addition to its polymerase activity. After base removal by a glycosylase and backbone cleavage by an AP endonuclease, these polymerases remove the abasic site (a -deoxyribose phosphate) and fill in the very short gap. The frequency of mutation due to DNA polymerase activity is minimized by the short lengths (often one nucleotide) of DNA synthesized. Mutations are genomic changes that alter the information in DNA. When mutations occur in genes encoding enzymes involved in DNA repair, the loss of function can lead to cancer.
Mutations are genomic changes that alter the information in DNA. When mutations occur in genes encoding enzymes involved in DNA repair, the loss of function can lead to cancer.