28.3 Regulation of Gene Expression in Eukaryotes
Initiation of transcription is a crucial regulation point for gene expression in all organisms. Although eukaryotes and bacteria use some of the same regulatory mechanisms, the regulation of transcription in the two systems is fundamentally different.
We can define a transcriptional ground state as the inherent activity of promoters and transcriptional machinery in vivo in the absence of regulatory sequences. In bacteria, RNA polymerase generally has access to every promoter and can bind and initiate transcription at some level of efficiency in the absence of activators or repressors. In eukaryotes, however, strong promoters are generally inactive in vivo in the absence of regulatory proteins. This fundamental difference gives rise to at least five important features that distinguish the regulation of gene expression at eukaryotic promoters from that observed in bacteria.
 We can define a transcriptional ground state as the inherent activity of promoters and transcriptional machinery in vivo in the absence of regulatory sequences. In bacteria, RNA polymerase generally has access to every promoter and can bind and initiate transcription at some level of efficiency in the absence of activators or repressors. In eukaryotes, however, strong promoters are generally inactive in vivo in the absence of regulatory proteins. This fundamental difference gives rise to at least five important features that distinguish the regulation of gene expression at eukaryotic promoters from that observed in bacteria.
We can define a transcriptional ground state as the inherent activity of promoters and transcriptional machinery in vivo in the absence of regulatory sequences. In bacteria, RNA polymerase generally has access to every promoter and can bind and initiate transcription at some level of efficiency in the absence of activators or repressors. In eukaryotes, however, strong promoters are generally inactive in vivo in the absence of regulatory proteins. This fundamental difference gives rise to at least five important features that distinguish the regulation of gene expression at eukaryotic promoters from that observed in bacteria.First, access to eukaryotic promoters is restricted by the structure of chromatin, and activation of transcription is associated with many changes in chromatin structure in the transcribed region. Second, although eukaryotic cells have both positive and negative regulatory mechanisms, positive mechanisms are more prominent. Almost every eukaryotic gene requires activation to be transcribed. Third, regulatory mechanisms involving lncRNAs are more common in eukaryotic transcriptional regulation. Fourth, eukaryotic cells have larger, more complex multimeric regulatory proteins than do bacteria. Finally, transcription in the eukaryotic nucleus is separated from translation in the cytoplasm in both space and time.
The complexity of regulatory circuits in eukaryotic cells is extraordinary, as is evident from the following discussion. The section ends with an illustrated description of one of the most elaborate circuits: the regulatory cascade that controls development in fruit flies.
Transcriptionally Active Chromatin Is Structurally Distinct from Inactive Chromatin
The effects of chromosome structure on gene regulation in eukaryotes have no clear parallel in bacteria. In the eukaryotic cell cycle, interphase chromosomes appear, at first viewing, to be dispersed and amorphous (see Fig. 24-22). Nevertheless, several forms of chromatin can be found along these chromosomes. About 10% of the chromatin in a typical eukaryotic cell is in a more condensed form than the rest of the chromatin. This form, heterochromatin, is transcriptionally inactive. Heterochromatin is generally associated with particular chromosome structures — the centromeres, for example. The remaining, less condensed chromatin is called euchromatin.
Transcription of a eukaryotic gene is strongly repressed when its DNA is condensed within heterochromatin. Some, but not all, of the euchromatin is transcriptionally active. Transcriptionally active chromosomal regions are distinguished from heterochromatin in at least three ways: the positioning of nucleosomes, the presence of histone variants, and the covalent modification of nucleosomes. These transcription-associated structural changes in chromatin are collectively called chromatin remodeling. The remodeling employs a set of enzymes that promote these changes (Table 28-2).
Enzyme complexa |
Oligomeric structure (number of polypeptides) |
Source |
Activities |
|---|---|---|---|
Histone movement, replacement, or editing, requiring ATP |
|||
SWI/SNF family |
Eukaryotes |
Nucleosome remodeling; transcriptional activation |
|
ISWI family |
2−4 |
Eukaryotes |
Nucleosome remodeling; transcriptional repression; transcriptional activation in some cases |
CHD family |
1−10 |
Eukaryotes |
Nucleosome remodeling; nucleosome ejection for transcriptional activation; some have repressive roles |
INO80 family |
Eukaryotes |
Nucleosome remodeling; transcriptional activation; family member SWR1 engages in replacement of H2A-H2B with H2AZ-H2B |
|
Histone modification |
|||
GCN5-ADA2-ADA3 |
3 |
Yeast |
GCN5 has type A HAT activity |
SAGA/PCAF |
Eukaryotes |
Includes GCN5-ADA2-ADA3; acetylates residues in H3, H2B, H2AZ |
|
NuA4 |
Eukaryotes |
EsaI component has HAT activity; acetylates H4, H2A, and H2AZ |
|
Histone chaperones not requiring ATP |
|||
HIRA |
1 |
Eukaryotes |
Deposition of H3.3 during transcription |
a The abbreviations for eukaryotic genes and proteins are often more confusing or obscure than those used for bacteria. SWI (switching) was discovered as a protein required for expression of certain genes involved in mating-type switching in yeast, and SNF (sucrose nonfermenting) as a factor for expression of the yeast gene for sucrase. Subsequent studies revealed multiple SWI and SNF proteins that act in a complex. The SWI/SNF complex has a role in expression of a wide range of genes and has been found in many eukaryotes, including humans. ISWI is imitation SWI. CHD is chromodomain, helicase, DNA binding; INO80 is inositol-requiring 80; and SWR1 is SWi2/Snf2-related ATPase 1. The complex of GCN5 (general control nonderepressible) and ADA (alteration/deficiency in activation) proteins was discovered during investigation of the regulation of nitrogen metabolism genes in yeast. These proteins can be part of the larger SAGA (SPF, ADA2,3, GCN5, acetyltransferase) complex in yeasts. The equivalent of SAGA in humans is PCAF (p300/CBP-associated factor). NuA4 is nucleosome acetyltransferase of H4; ESA1 is essential SAS2-related acetyltransferase; HIRA is histone regulator A. |
|||
Four known families of chromatin remodeling complexes, distinguished by their structural features, act directly to alter nucleosome composition in transcribed regions. They may unwrap, translocate, remove, or exchange nucleosomes on the DNA, hydrolyzing ATP in the process (Table 28-2; see the table footnote for an explanation of the abbreviated names of enzyme complexes described here). In some cases, the enzymes catalyze the exchange of pairs of histones within nucleosomes to alter nucleosome composition. The multitude of different complexes are specialized to function at particular genes or chromosomal regions. There are two related complexes in the SWI/SNF family in all eukaryotic cells, both of which remodel chromatin so that nucleosomes are ejected from the DNA near transcription start sites. They appear to be involved in a dynamic cycle to allow replacement of nucleosomes with transcription factors (Fig. 28-28). The two distinct complexes generally function at different sets of genes. Most of the ISWI family complexes optimize nucleosome spacing to allow chromatin assembly and transcriptional silencing. There are generally 9 or 10 different CHD family complexes in eukaryotic cells, separated into three subfamilies. The different family members have specialized roles, either ejecting nucleosomes to activate transcription or assembling chromatin to repress transcription. The INO80 family complexes have a variety of roles in remodeling chromatin for transcriptional activation and DNA repair. One family member, SWR1, promotes subunit exchange in nucleosomes to introduce histone variants such as H2AZ (see Box 24-1), found in transcriptionally active regions.
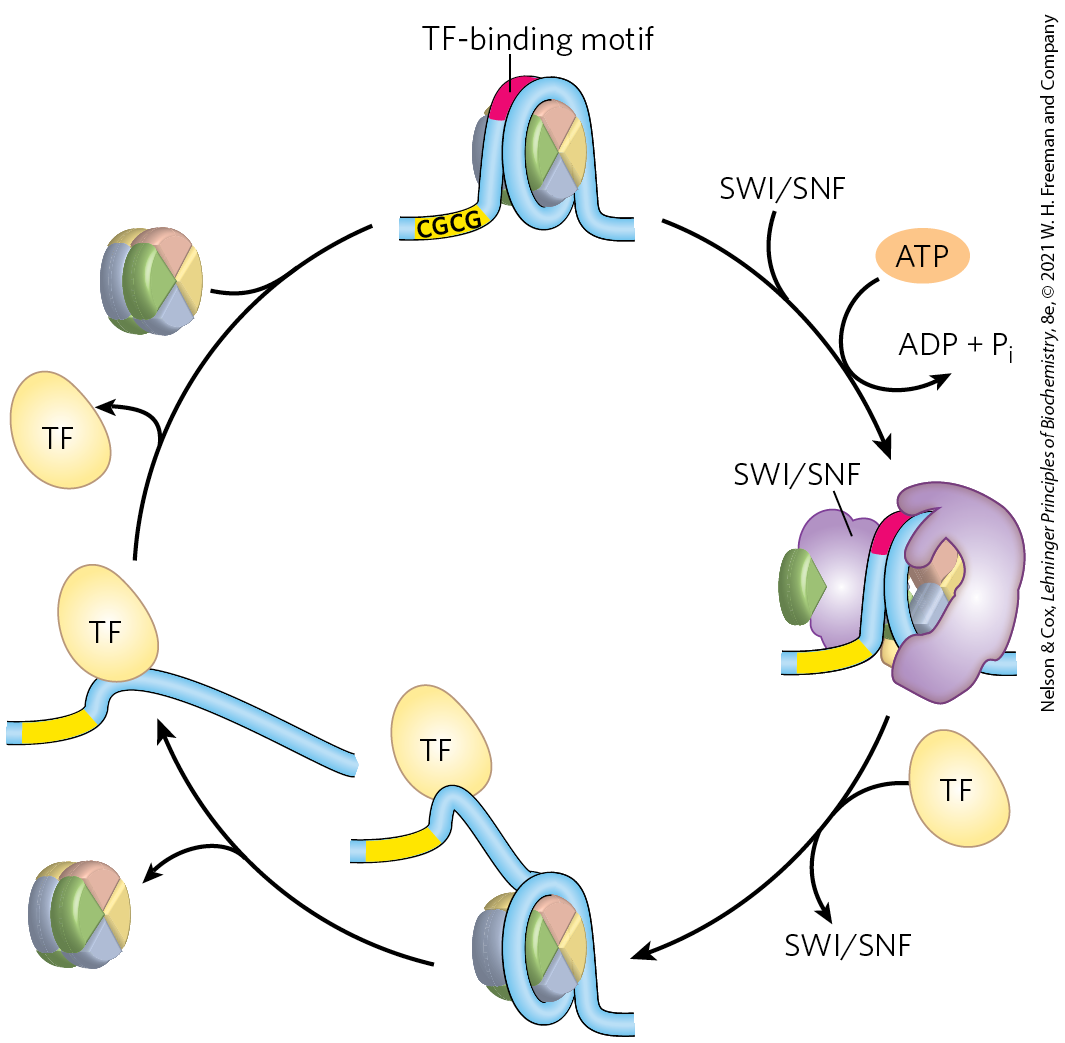
FIGURE 28-28 Nucleosome ejection by a SWI/SNF remodeler. The SWI/SNF enzyme engulfs the nucleosome, interacting with short CGCG sequences nearby. With the aid of ATP hydrolysis, the DNA is partially separated from the nucleosome, exposing a site for transcription factor (TF) binding. After the transcription factor is bound, the nucleosome is ejected. When transcription is no longer needed, the nucleosome can again replace the transcription factor or factors, completing the cycle. [Information from S. Brahma and S. Henikoff, Trends Biochem. Sci. 45:13, 2020.]
The covalent modification of histones is altered dramatically within transcriptionally active chromatin. The core histones of nucleosome particles (H2A, H2B, H3, H4; see Fig. 24-24) are modified by methylation of Lys or Arg residues, phosphorylation of Ser or Thr residues, acetylation (see below), ubiquitination (see Fig. 27-47), or SUMOylation (SUMOs are small ubiquitin-like modifiers). Each of the core histones has two distinct structural domains. A central domain is involved in histone-histone interaction and the wrapping of DNA around the nucleosome. A lysine-rich amino-terminal domain is generally positioned near the exterior of the assembled nucleosome particle; the covalent modifications occur at specific residues concentrated in this amino-terminal domain. The patterns of modification have led some researchers to propose the existence of a histone code, in which modification patterns are recognized by enzymes that alter the structure of chromatin. Indeed, some of the modifications are essential for interactions with proteins that play key roles in transcription.
The acetylation and methylation of histones figure prominently in the processes that activate chromatin for transcription. During transcription, histone H3 in nucleosomes is methylated (by specific histone methylases) at near the end of the coding region and at within the coding region. These methylations enable the binding of histone acetyltransferases (HATs), enzymes that acetylate particular Lys residues. Cytosolic (type B) HATs acetylate newly synthesized histones before the histones are imported into the nucleus. The subsequent assembly of the histones into chromatin after replication is facilitated by histone chaperones: CAF1 for H3 and H4 (see Box 24-1), and NAP1 for H2A and H2B.
Where chromatin is being activated for transcription, the nucleosomal histones are further acetylated by nuclear (type A) HATs. The acetylation of multiple Lys residues in the amino-terminal domains of histones H3 and H4 can reduce the affinity of the entire nucleosome for DNA. Acetylation of particular Lys residues is critical for the interaction of nucleosomes with other proteins.
When transcription of a gene is no longer required, the extent of methylation and acetylation of nucleosomes in that vicinity is reduced as part of a general gene-silencing process that restores the chromatin to a transcriptionally inactive state. There are two known classes of demethylases. One class, called LSD (lysine-specific histone demethylases), first converts the linkage to an imine linkage, followed by hydrolysis to generate formaldehyde and the demethylated lysine. The other class of demethylases contains JmjC (Jumonji-C) domains, first hydroxylating the methyl group, which is again subsequently removed as formaldehyde. More than 20 JmjC domain–containing histone demethylases are encoded by mammalian genomes. They are part of the same α-ketoglutarate-dependent hydroxylase enzyme family that includes the enzyme that hydroxylates proline residues in collagen (see Box 4-2). These enzymes are strongly inhibited by 2-hydroxyglutarate, an unusual metabolite produced in abundance by a mutated form of isocitrate dehydrogenase that is common in human cancers (see Fig. 16-20). Within the tumors, the high levels of 2-hydroxyglutarate produce global changes in gene expression.
 When transcription of a gene is no longer required, the extent of methylation and acetylation of nucleosomes in that vicinity is reduced as part of a general gene-silencing process that restores the chromatin to a transcriptionally inactive state. There are two known classes of demethylases. One class, called LSD (lysine-specific histone demethylases), first converts the linkage to an imine linkage, followed by hydrolysis to generate formaldehyde and the demethylated lysine. The other class of demethylases contains JmjC (Jumonji-C) domains, first hydroxylating the methyl group, which is again subsequently removed as formaldehyde. More than 20 JmjC domain–containing histone demethylases are encoded by mammalian genomes. They are part of the same α-ketoglutarate-dependent hydroxylase enzyme family that includes the enzyme that hydroxylates proline residues in collagen (see
When transcription of a gene is no longer required, the extent of methylation and acetylation of nucleosomes in that vicinity is reduced as part of a general gene-silencing process that restores the chromatin to a transcriptionally inactive state. There are two known classes of demethylases. One class, called LSD (lysine-specific histone demethylases), first converts the linkage to an imine linkage, followed by hydrolysis to generate formaldehyde and the demethylated lysine. The other class of demethylases contains JmjC (Jumonji-C) domains, first hydroxylating the methyl group, which is again subsequently removed as formaldehyde. More than 20 JmjC domain–containing histone demethylases are encoded by mammalian genomes. They are part of the same α-ketoglutarate-dependent hydroxylase enzyme family that includes the enzyme that hydroxylates proline residues in collagen (see 
Histone acetylation is reduced by the action of histone deacetylases (HDACs). The deacetylases include SIRT1, SIRT2, SIRT6, and SIRT7, which are -dependent enzymes in the sirtuin family (SIRT1–7 in mammals). These enzymes deacetylate specific Lys residues in histones and other, cytoplasmic targets. In addition to the removal of certain acetyl groups, new covalent modification of histones marks chromatin as transcriptionally inactive. For example, of histone H3 is often methylated in heterochromatin.
The net effect of chromatin remodeling in the context of transcription is to make a segment of the chromosome more accessible and to “label” (chemically modify) it so as to facilitate the binding and activity of transcription factors that regulate expression of the gene or genes in that region.
Most Eukaryotic Promoters Are Positively Regulated
As already noted, eukaryotic RNA polymerases have little or no intrinsic affinity for their promoters. The default state of eukaryotic genes is “off,” and initiation of transcription is almost always dependent on the action of multiple activator proteins. One important reason for the apparent predominance of positive regulation seems obvious: the storage of DNA within chromatin effectively renders most promoters inaccessible, so genes are silent in the absence of other regulation. The structure of chromatin affects access to some promoters more than others, but repressors that bind to DNA so as to preclude access of RNA polymerase (negative regulation) would often be simply redundant. Other factors must be at play in the use of positive regulation, and speculation generally centers around two: the large size of eukaryotic genomes and the greater efficiency of positive regulation.
First, nonspecific DNA binding of regulatory proteins becomes a more important problem in the much larger genomes of higher eukaryotes. And the chance that a single specific binding sequence will occur randomly at an inappropriate site also increases with genome size. Combinatorial control thus becomes important in a large genome (Fig. 28-29). Specificity for transcriptional activation can be improved if each of several positive regulatory proteins must bind specific DNA sequences to activate a gene. The average number of regulatory sites for a gene in a multicellular organism is six, and genes that are regulated by a dozen such sites are common. The requirement for binding of several positive regulatory proteins to specific DNA sequences vastly reduces the probability of the random occurrence of a functional juxtaposition of all the necessary binding sites. This requirement also reduces the number of regulatory proteins that must be encoded by a genome to regulate all of its genes (Fig. 28-28). Thus, a new regulator is not needed for every gene, although regulation is complex enough in higher eukaryotes that regulatory proteins may represent 5% to 10% of all protein-coding genes.
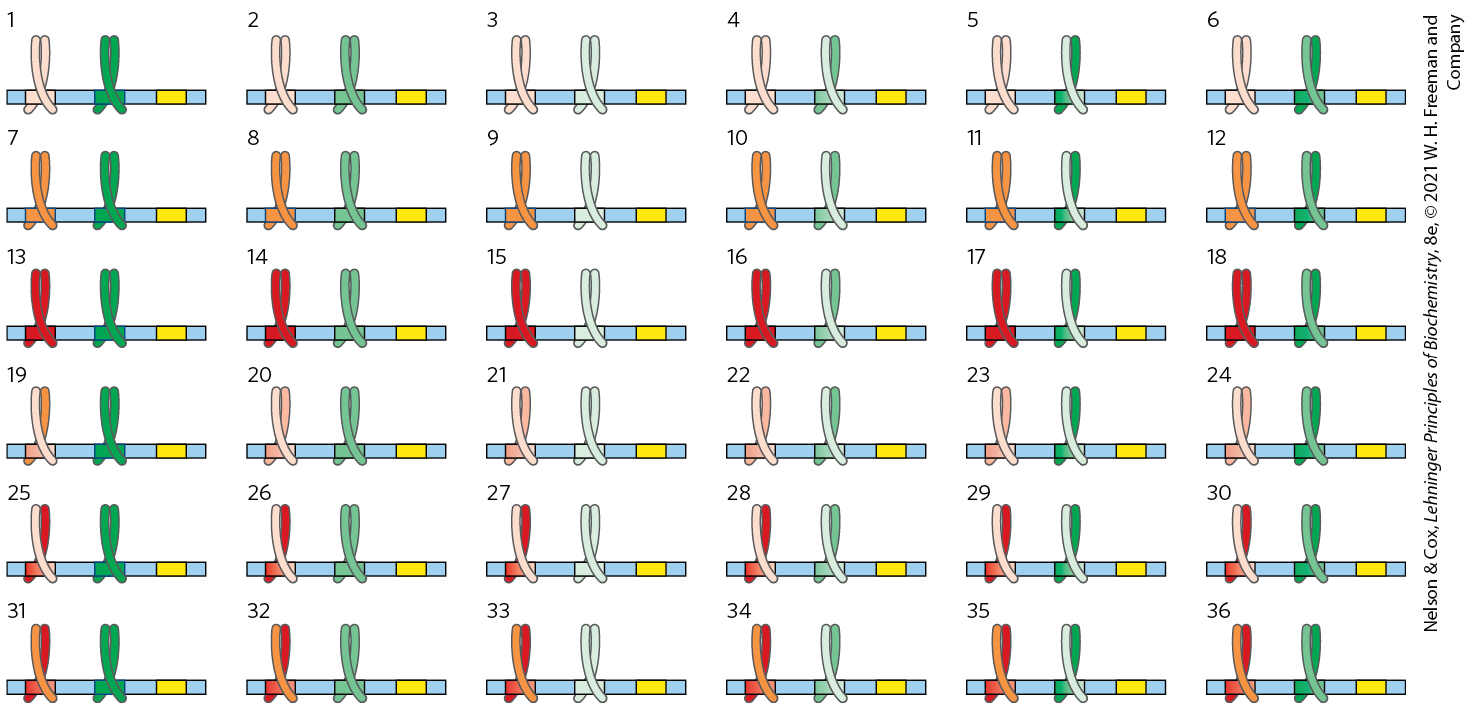
FIGURE 28-29 The advantages of combinatorial control. Combinatorial control allows specific regulation of many genes using a limited repertoire of regulatory proteins. Consider the possibilities inherent in regulation by two different families of leucine zipper proteins (red and green). If each regulatory gene family had three members (as shown here, in dark, medium, and light shades, each binding to a different DNA sequence) that could freely form either homo- or heterodimers, there would be six possible dimeric species in each family and each dimer would recognize a different bipartite regulatory DNA sequence. If a gene had a regulatory site for each protein family, 36 different regulatory combinations would be possible, using just the six proteins from these two families. With six or more sites used in the regulation of a typical eukaryotic gene, the number of possible variants is much greater than this example suggests.
In principle, a similar combinatorial strategy could be used by multiple negative regulatory elements, but this brings us to the second reason for the use of positive regulation: it is simply more efficient. If the ~20,000 genes in the human genome were negatively regulated, each cell would have to synthesize, at all times, all of the different repressors in concentrations sufficient to permit specific binding to each “unwanted” gene. In positive regulation, most of the genes are usually inactive (that is, RNA polymerases do not bind to the promoters) and the cell synthesizes only the activator proteins needed to promote transcription of the subset of genes required in the cell at that time.
These arguments notwithstanding, there are examples of negative regulation in eukaryotes, from yeasts to humans, as we shall see. Some of that negative regulation involves lncRNAs, which are more economical to synthesize than repressor proteins.
DNA-Binding Activators and Coactivators Facilitate Assembly of the Basal Transcription Factors
To continue our exploration of the regulation of gene expression in eukaryotes, we return to the interactions between promoters and RNA polymerase II (Pol II), the enzyme responsible for the synthesis of eukaryotic mRNAs. Although many (but not all) Pol II promoters include the TATA box and Inr (initiator) sequences, with their standard spacing (see Fig. 26-8), they vary greatly in both the number and the location of additional sequences required for the regulation of transcription.
The additional regulatory sequences, generally bound by transcription activators, are usually called enhancers in higher eukaryotes and upstream activator sequences (UASs) in yeast. A typical enhancer may be found hundreds or even thousands of base pairs upstream from the transcription start site, or may even be downstream, within the gene itself. When bound by the appropriate regulatory proteins, an enhancer increases transcription at nearby promoters regardless of its orientation in the DNA. The UASs of yeast function in a similar way, although generally they must be positioned upstream and within a few hundred base pairs of the transcription start site.
Successful binding of the active Pol II holoenzyme at one of its promoters usually requires the combined action of proteins of five types: (1) transcription activators, which bind to enhancers or UASs and facilitate transcription; (2) architectural regulators, which facilitate DNA looping; (3) chromatin modification and remodeling proteins, described above; (4) coactivators; and (5) basal transcription factors, also called general transcription factors (see Fig. 26-9, Table 26-2), required at most Pol II promoters (Fig. 28-30). The coactivators are required for essential communication between activators and the complex composed of Pol II and the basal transcription factors. Coactivators also play a direct role in assembly of the preinitiation complex (PIC). Furthermore, a variety of repressor proteins can interfere with communication between Pol II and the activators, resulting in repression of transcription (Fig. 28-30b). Here we focus on the protein complexes shown in Figure 28-30 and how they interact to activate transcription.
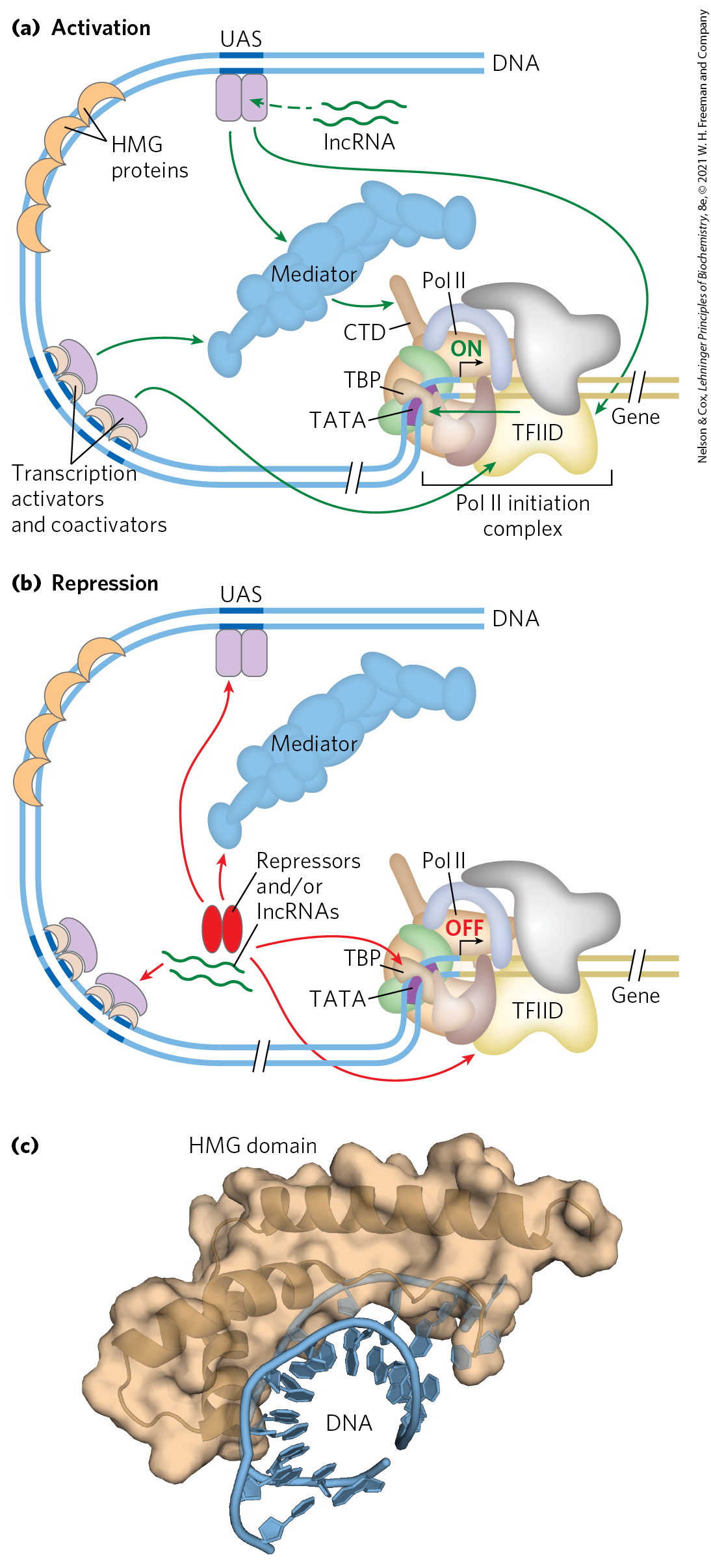
FIGURE 28-30 Eukaryotic promoters and regulatory proteins. RNA polymerase II and its associated basal (general) transcription factors form a preinitiation complex at the TATA box and Inr site of the cognate promoters, a process facilitated by transcription activators, acting through coactivators (Mediator, TFIID, or both). (a) A composite promoter with typical sequence elements and protein complexes found in both yeast and higher eukaryotes. The carboxyl-terminal domain (CTD) of Pol II (see Fig. 26-9) is an important point of interaction with Mediator and other protein complexes. Histone modification enzymes (not shown) catalyze methylation and acetylation; remodeling enzymes alter the content and placement of nucleosomes. The transcription activators have distinct DNA-binding domains and activation domains. In some cases, their function is affected by interaction with lncRNAs. Arrows indicate common modes of interaction often required for the activation of transcription. The HMG proteins are a common type of architectural regulator (see Fig. 28-5), allowing the looping of the DNA required to bring together system components bound at distant binding sites. (b) Eukaryotic transcriptional repressors function through a range of mechanisms. Some bind directly to DNA, displacing a protein complex required for activation (not shown); many others interact with various parts of the transcription or activation protein complexes to prevent activation. Possible points of interaction are indicated with arrows. (c) The structure of an HMG protein complex with DNA shows how HMG proteins facilitate DNA looping. The binding is relatively nonspecific, although DNA sequence preferences have been identified for many HMG proteins. Shown here is the HMG domain of the protein HMG-D of Drosophila, bound to DNA. [(c) Data from PDB ID 1QRV, F. V. Murphy IV et al., EMBO J. 18:6610, 1999.]
Transcription Activators
The requirements for activators vary greatly from one promoter to another. A few are known to activate transcription at hundreds of promoters, whereas others are specific for a few promoters. Many activators are sensitive to the binding of signal molecules, providing the capacity to activate or deactivate transcription in response to a changing cellular environment. Some enhancers bound by activators are quite distant from the promoter’s TATA box. Multiple enhancers (often six or more) are bound by a similar number of activators for a typical gene, providing combinatorial control and response to multiple signals.
Some transcription activators can bind to both DNA and RNA, and their function is affected by one or more lncRNAs. The protein , for example (Fig. 28-14), activates transcription of many genes involved in the immune response and cytokine production. It can bind to a DNA enhancer site or, alternatively, to an lncRNA called lethe, named after the river of forgetfulness in Greek mythology. The lncRNA reduces transcription of genes controlled by .
Architectural Regulators
How do activators function at a distance? The answer in most cases seems to be that, as indicated earlier, the intervening DNA is looped so that the various protein complexes can interact directly. The looping is promoted by architectural regulators that are abundant in chromatin and bind to DNA with limited specificity. Most prominently, the high mobility group (HMG) proteins (Fig. 28-29c; “high mobility” refers to their electrophoretic mobility in polyacrylamide gels) play an important structural role in chromatin remodeling and transcriptional activation.
Coactivator Protein Complexes
Most transcription requires the presence of additional protein complexes. Some major regulatory protein complexes that interact with Pol II have been defined both genetically and biochemically. These coactivator complexes act as intermediaries between the transcription activators and the Pol II complex.
Mediator, a complex consisting of 25 (yeast) to 30 (human) polypeptides, is a major eukaryotic coactivator (Fig. 28-30). Many of the 25 core polypeptides are highly conserved from fungi to humans. A subcomplex of four subunits has a kinase role, interacting transiently with the remainder of the Mediator complex, and may dissociate prior to transcription initiation. Mediator binds tightly to the carboxyl-terminal domain (CTD) of the largest subunit of Pol II. The Mediator complex is required for both basal and regulated transcription at many promoters used by Pol II, and it also stimulates phosphorylation of the CTD by TFIIH (a basal transcription factor). Transcription activators interact with one or more components of the Mediator complex, with the precise interaction sites differing from one activator to another. Coactivator complexes function at or near the promoter’s TATA box.
Additional coactivators, functioning with one or a few genes, have also been described. Some of these operate in conjunction with Mediator, and some may act in systems that do not employ Mediator.
TATA-Binding Protein and Basal Transcription Factors
The first component to bind in the assembly of a preinitiation complex (PIC) at the TATA box of a typical Pol II promoter is the TATA-binding protein (TBP). At promoters lacking a TATA box, TBP is usually delivered as part of a larger complex (13 to 14 subunits) called TFIID. The complete complex also includes the basal transcription factors TFIIB, TFIIE, TFIIF, TFIIH; Pol II; and perhaps TFIIA. This minimal PIC, however, is often insufficient for initiation of transcription and generally does not form at all if the promoter is obscured within chromatin. Positive regulation, leading to transcription, is imposed by the activators and coactivators. Mediator interacts directly with TFIIH and TFIIE, allowing their recruitment to the PIC.
Choreography of Transcriptional Activation
We can now begin to piece together the sequence of transcriptional activation events at a typical Pol II promoter (Fig. 28-31). The exact order of binding of some components may vary, but the model in Figure 28-31 illustrates the principles of activation as well as one common path. Many transcription activators have significant affinity for their binding sites even when the sites are within condensed chromatin. The binding of activators is often the event that triggers subsequent activation of the promoter. Binding of one activator may enable the binding of others, gradually displacing some nucleosomes.
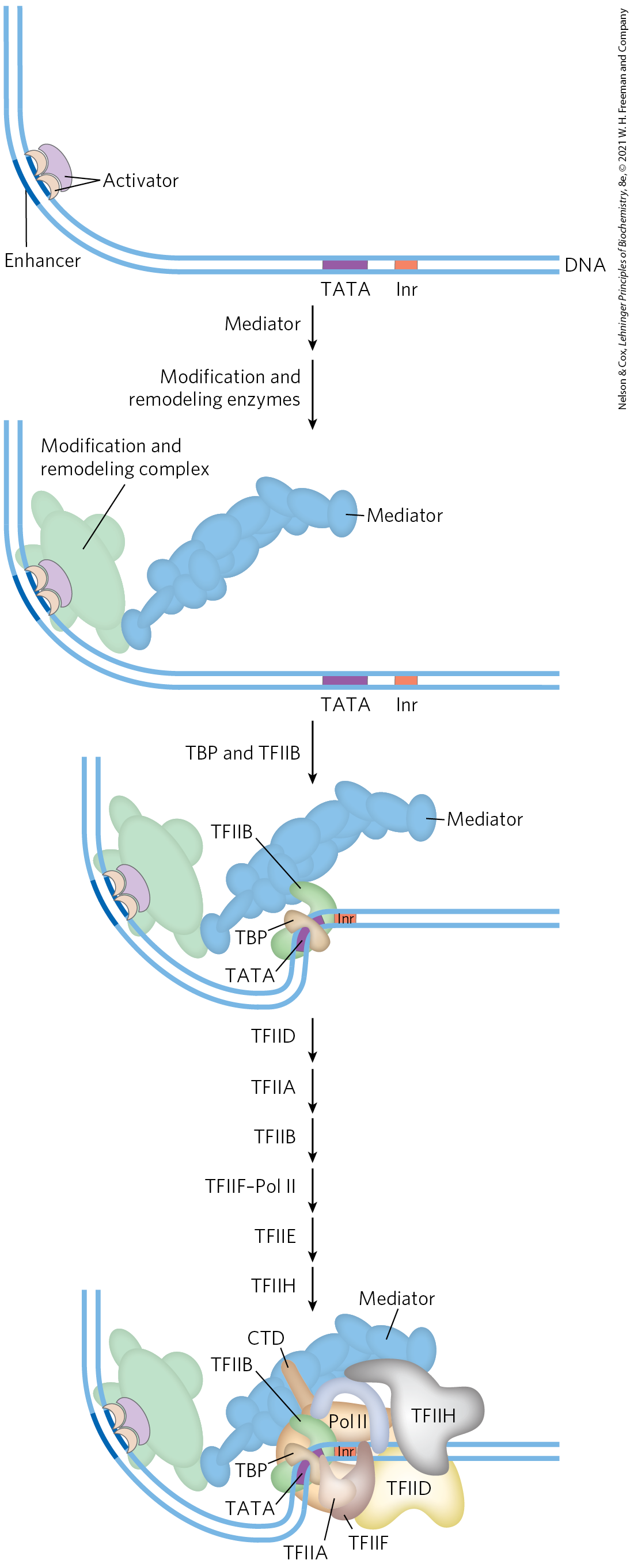
FIGURE 28-31 The components of transcriptional activation. Activators bind the DNA first. The activators recruit the histone modification/nucleosome remodeling complexes and a coactivator such as Mediator. Mediator facilitates the binding of TBP (or TFIID) and TFIIB, and the other basal transcription factors and Pol II then bind. Phosphorylation of the CTD of Pol II leads to transcription initiation (not shown). [Information from J. A. D’Alessio et al., Mol. Cell 36:924, 2009.]
Crucial remodeling of the chromatin then takes place in stages, facilitated by interactions between activators and HATs or enzyme complexes such as SWI/SNF, or both. In this way, a bound activator can draw in other components necessary for further chromatin remodeling to permit transcription of specific genes. The bound activators interact with the large Mediator complex. Mediator, in turn, provides an assembly surface for the binding of, first, TBP (or TFIID), then TFIIB, and then other components of the PIC, including Pol II. Mediator stabilizes the binding of Pol II and its associated transcription factors and greatly facilitates formation of the PIC. Complexity in these regulatory circuits is the rule rather than the exception, with multiple DNA-bound activators promoting transcription.
The script can change from one promoter to another. For example, many promoters have a different set of recognition sequences and may not have a TATA box, and in multicellular eukaryotes the subunit composition of factors such as TFIID can vary from one tissue to another. However, most promoters seem to require a precisely ordered assembly of components to initiate transcription. The assembly process is not always fast. For some genes it may take minutes; for certain genes of higher eukaryotes, the process can take days.
The Genes of Galactose Metabolism in Yeast Are Subject to Both Positive and Negative Regulation
Some of the general principles described above can be illustrated by one well-studied eukaryotic regulatory circuit (Fig. 28-32). The enzymes required for the importation and metabolism of galactose in yeast are encoded by genes scattered over several chromosomes (Table 28-3). Each of the GAL genes is transcribed separately, and yeast cells have no operons like those in bacteria. However, all the GAL genes have similar promoters and are regulated coordinately by a common set of proteins. The promoters for the GAL genes consist of the TATA box and Inr sequences, as well as an upstream activator sequence recognized by the transcription activator Gal4 protein (Gal4p). Regulation of gene expression by galactose entails an interplay between Gal4p and two other proteins, Gal80p and Gal3p. Gal80p forms a complex with Gal4p, preventing Gal4p from functioning as an activator of the GAL promoters. When galactose is present, it binds Gal3p, which then interacts with the Gal80p-Gal4p complex and allows Gal4p to function as an activator at the GAL promoters. As the various galactose genes are induced and their products build up, Gal3p may be replaced with Gal1p (a galactokinase needed for galactose metabolism that also acts as a regulator) for sustained activation of the regulatory circuit.
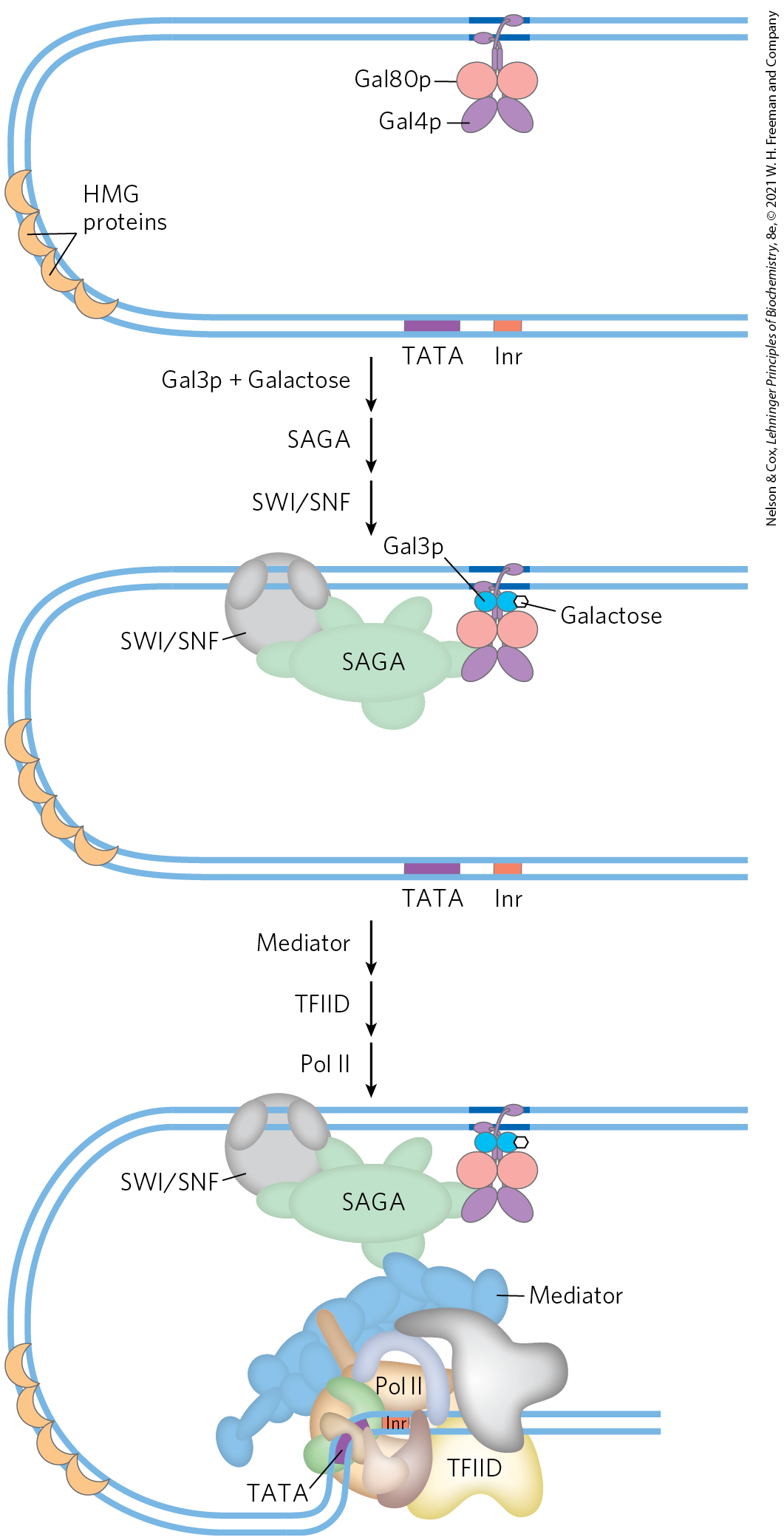
FIGURE 28-32 Regulation of transcription of GAL genes in yeast. Galactose imported into the yeast cell is converted to glucose 6-phosphate by a pathway involving five enzymes, whose genes are scattered over three chromosomes (see Table 28-3). Transcription of these genes is regulated by the combined actions of the proteins Gal4p, Gal80p, and Gal3p, with Gal4p playing the central role of transcription activator. The Gal4p-Gal80p complex is inactive. Binding of galactose to Gal3p leads to interaction of Gal3p with the Gal80p-Gal4p complex and activates Gal4p. The Gal4p subsequently recruits SAGA, Mediator, and TFIID to the galactose promoters, leading to recruitment of RNA polymerase II and initiation of transcription. Chromatin remodeling to allow transcription also requires a SWI/SNF complex.
Relative protein expression in different carbon sources |
||||||
|---|---|---|---|---|---|---|
Gene |
Protein function |
Chromosomal location |
Protein size (number of residues) |
Glucose |
Glycerol |
Galactose |
Regulated genes |
||||||
GAL1 |
Galactokinase |
II |
528 |
|||
GAL2 |
Galactose permease |
XII |
574 |
|||
PGM2 |
Phosphoglucomutase |
XIII |
569 |
|||
GAL7 |
Galactose 1-phosphate uridylyltransferase |
II |
365 |
|||
GAL10 |
UDP-glucose 4-epimerase |
II |
699 |
|||
MEL1 |
α-Galactosidase |
II |
453 |
|||
Regulatory genes |
||||||
GAL3 |
Inducer |
IV |
520 |
|||
GAL4 |
Transcriptional activator |
XVI |
881 |
|||
GAL80 |
Transcriptional inhibitor |
XIII |
435 |
|||
Information from R. Reece and A. Platt, Bioessays 19:1001, 1997. |
||||||
Other protein complexes also have a role in activating transcription of the GAL genes. These include the SAGA complex for histone acetylation and chromatin remodeling, the SWI/SNF complex for chromatin remodeling, and Mediator. The Gal4 protein is responsible for recruitment of these additional factors needed for transcriptional activation. SAGA may be the first and primary recruitment target for Gal4p.
Glucose is the preferred carbon source for yeast, as it is for bacteria. When glucose is present, most of the GAL genes are repressed — whether galactose is present or not. The GAL regulatory system described above is effectively overridden by a complex catabolite repression system that includes several proteins (not depicted in Fig. 28-32).
Transcription Activators Have a Modular Structure
Transcription activators typically have a distinct structural domain for specific DNA binding and one or more additional domains for transcriptional activation or for interaction with other regulatory proteins. Interaction of two regulatory proteins is often mediated by domains containing leucine zippers (Fig. 28-15) or helix-loop-helix motifs (Fig. 28-16). We consider here three distinct types of structural domains used in activation by the transcription activators Gal4p, Sp1, and CTF1 (Fig. 28-33a).
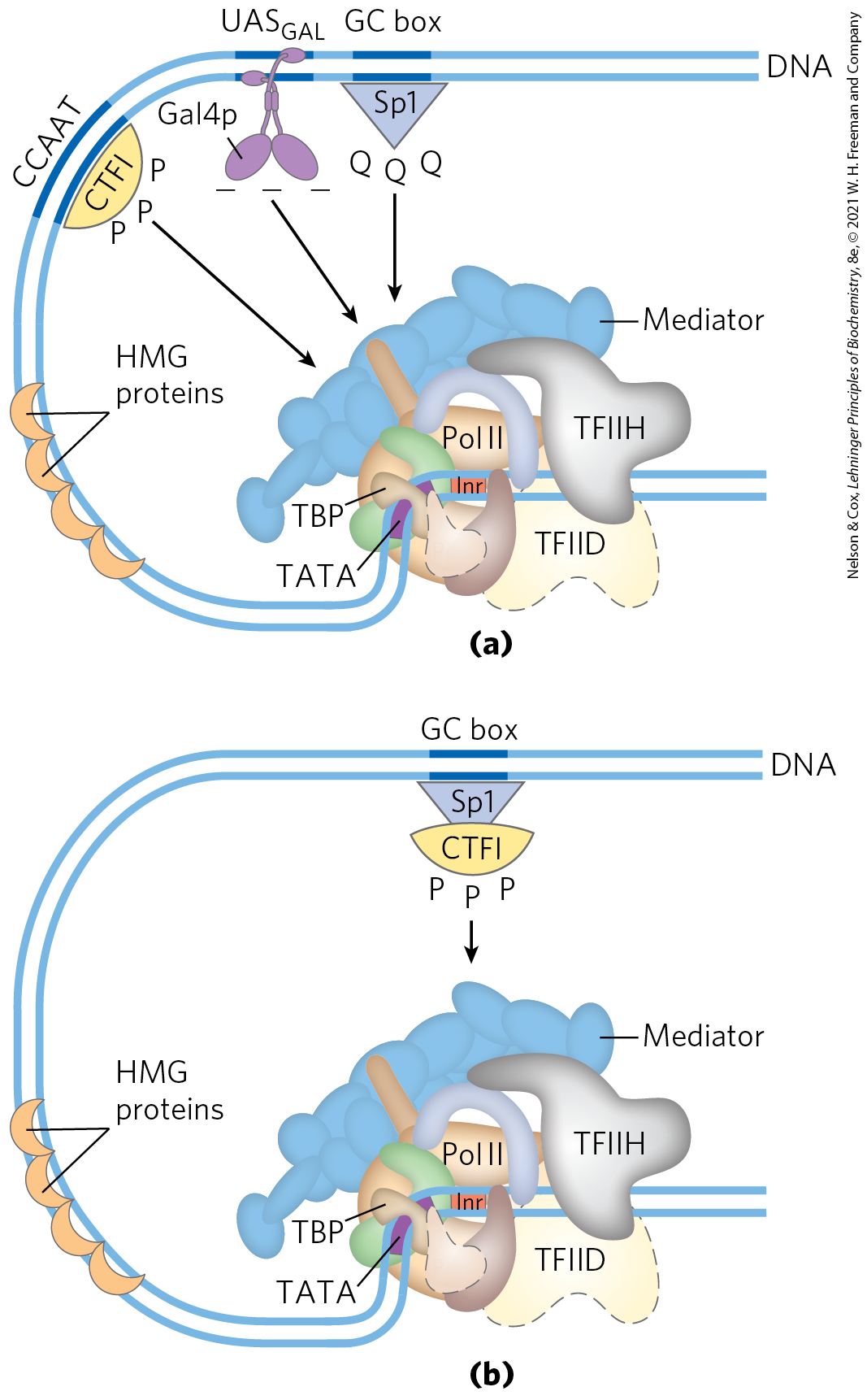
FIGURE 28-33 Transcription activators. (a) Typical activators such as CTF1, Gal4p, and Sp1 have a DNA-binding domain and an activation domain. The nature of the activation domain is indicated by symbols: – – –, acidic; Q Q Q, glutamine-rich; P P P, proline-rich. These proteins generally activate transcription by interacting with coactivator complexes such as Mediator. Note that the binding sites illustrated here are not generally found together near a single gene. (b) A chimeric protein containing the DNA-binding domain of Sp1 and the activation domain of CTF1 activates transcription if a GC box is present.
Gal4p contains a zinc finger–like structure in its DNA-binding domain, near the amino terminus; this domain has six Cys residues that coordinate two . The protein functions as a homodimer (with dimerization mediated by interactions between two coiled coils) and binds to , a palindromic DNA sequence about 17 bp long. Gal4p has a separate activation domain with many acidic amino acid residues. Experiments that substitute a variety of different peptide sequences for the acidic activation domain of Gal4p suggest that the acidic nature of this domain is critical to its function, although its precise amino acid sequence can vary considerably.
Sp1 is a transcription activator for many genes in higher eukaryotes. Its DNA-binding site, the GC box (consensus sequence GGGCGG), is usually quite near the TATA box. The DNA-binding domain of the Sp1 protein is near its carboxyl terminus and contains three zinc fingers. Two other domains in Sp1 function in activation and are notable in that 25% of their amino acid residues are Gln. A wide variety of other activator proteins also have these glutamine-rich domains.
CTF1 (CCAAT-binding transcription factor 1) belongs to a family of transcription activators that bind a sequence called the CCAAT site (its consensus sequence is , where N is any nucleotide). The DNA-binding domain of CTF1 contains many basic amino acid residues, and the binding region is probably arranged as an α helix. This protein has neither a helix-turn-helix motif nor a zinc finger motif; its DNA-binding mechanism is not yet clear. CTF1 has a proline-rich activation domain, with Pro accounting for more than 20% of the amino acid residues.
The discrete activation and DNA-binding domains of regulatory proteins often act completely independently, as has been demonstrated in “domain-swapping” experiments. Genetic engineering techniques (Chapter 9) can join the proline-rich activation domain of CTF1 to the DNA-binding domain of Sp1 to create a protein that, like intact Sp1, binds to GC boxes on the DNA and activates transcription at a nearby promoter (as in Fig. 28-33b). The DNA-binding domain of Gal4p has similarly been replaced experimentally with the DNA-binding domain of the E. coli LexA repressor (of the SOS response; Fig. 28-21). This chimeric protein neither binds at nor activates the yeast GAL genes (as would intact Gal4p) unless the sequence in the DNA is replaced by the LexA recognition site.
Eukaryotic Gene Expression Can Be Regulated by Intercellular and Intracellular Signals
The effects of steroid hormones (and of thyroid and retinoid hormones, which have a similar mode of action) provide additional well-studied examples of the modulation of eukaryotic regulatory proteins by direct interaction with molecular signals (see Fig. 12-34). Unlike other types of hormones, steroid hormones do not have to bind to plasma membrane receptors. Instead, they can interact with intracellular receptors that are transcription activators. Steroid hormones too hydrophobic to dissolve readily in the blood (estrogen, progesterone, and cortisol, for example) travel on specific carrier proteins from their point of release to their target tissues. In the target tissue, the hormone passes through the plasma membrane by simple diffusion. Once inside the cell, the hormone interacts with one of two types of steroid-binding nuclear receptor (Fig. 28-34). In both cases, the hormone-receptor complex acts by binding to highly specific DNA sequences called hormone response elements (HREs), thereby altering gene expression. Acting at these sites, the receptors act as transcription activators, recruiting coactivators and Pol II (plus its associated transcription factors) to trigger transcription of the gene.
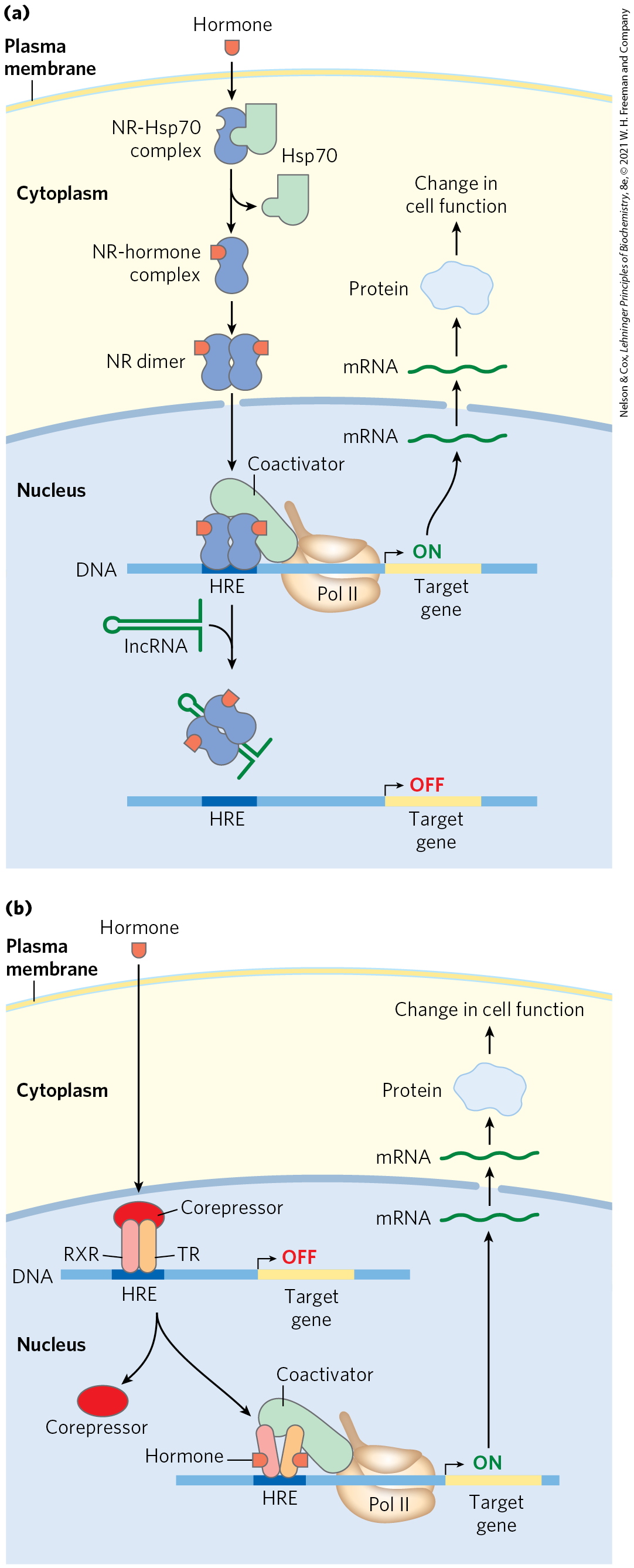
FIGURE 28-34 Mechanisms of steroid hormone receptor function. There are two types of steroid-binding nuclear receptors. (a) Monomeric type I receptors (NR) are found in the cytoplasm, in a complex with the heat shock protein Hsp70. Receptors for estrogen, progesterone, androgens, and glucocorticoids are of this type. When the steroid hormone binds, the Hsp70 dissociates and the receptor dimerizes, exposing a nuclear localization signal. The dimeric receptor, with hormone bound, migrates to the nucleus, where it binds to a hormone response element (HRE) and acts as a transcription activator. The activity of the receptor can be repressed by binding to an lncRNA (such as GAS5), which competes directly with binding to the HRE. (b) Type II receptors, by contrast, are always in the nucleus, bound to an HRE in the DNA and to a corepressor that renders the receptor inactive. The thyroid hormone receptor (TR) is of this type. The hormone migrates through the cytoplasm and diffuses across the nuclear membrane. In the nucleus it binds to a heterodimer consisting of the thyroid hormone receptor and the retinoid X receptor (RXR). A conformation change leads to dissociation of the corepressor, and the receptor then functions as a transcription activator.
The DNA sequences (HREs) to which hormone-receptor complexes bind are similar in length and arrangement for the various steroid hormones, but they differ in sequence. Each receptor has a consensus HRE sequence (Table 28-4) to which the hormone-receptor complex binds well, with each consensus consisting of two six-nucleotide sequences, either contiguous or separated by three nucleotides, in tandem or in a palindromic arrangement. The hormone receptors have a highly conserved DNA-binding domain with two zinc fingers (Fig. 28-35). The hormone-receptor complex binds to the DNA as a dimer, with the zinc finger domains of each monomer recognizing one of the six-nucleotide sequences. The ability of a given hormone to act through the hormone-receptor complex to alter the expression of a specific gene depends on the exact sequence of the HRE, its position relative to the gene, and the number of HREs associated with the gene.
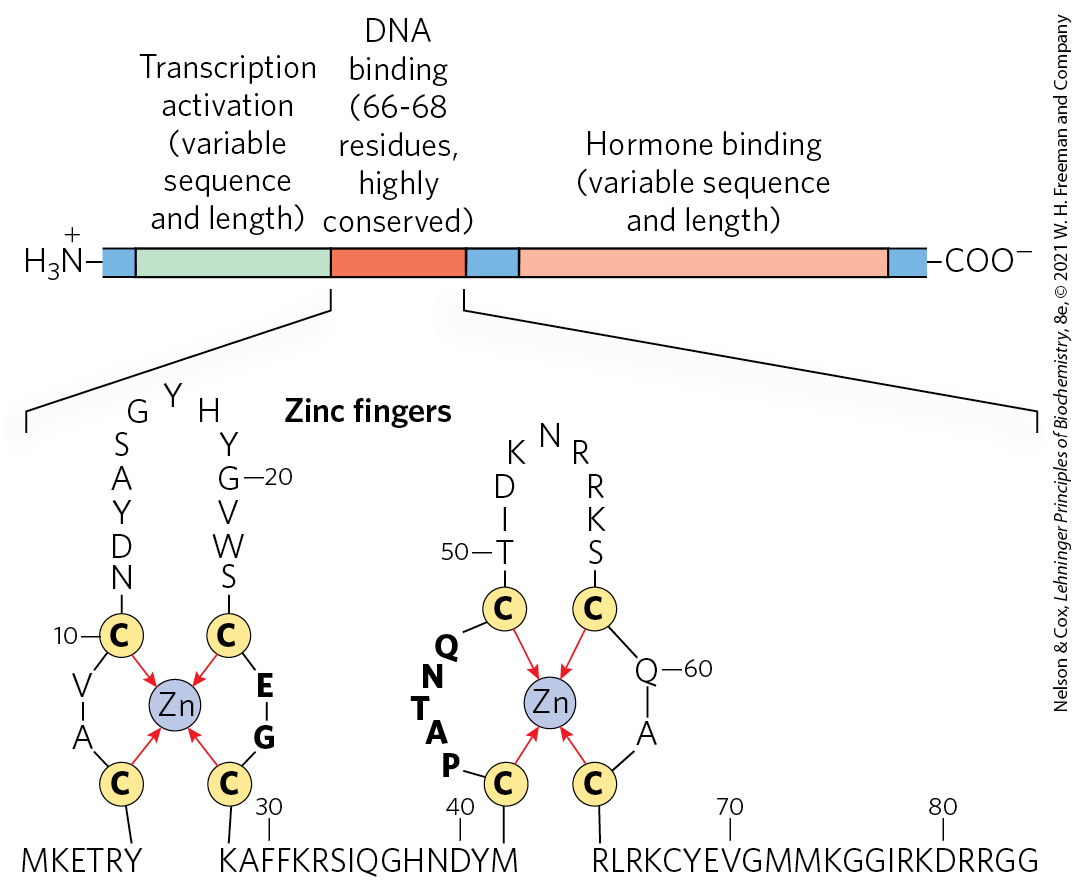
FIGURE 28-35 Typical steroid hormone receptors. These receptor proteins have a binding site for the hormone, a DNA-binding domain, and a region that activates transcription of the regulated gene. The highly conserved DNA-binding domain has two zinc fingers. The sequence shown here is that for the estrogen receptor, but the residues in bold type are common to all steroid hormone receptors.
Receptor |
HRE consensus sequence bounda |
|---|---|
Androgen |
|
Glucocorticoid |
|
Retinoic acid (some) |
|
Vitamin D |
|
Thyroid hormone |
|
RXb |
|
aN represents any nucleotide. bForms a dimer with the retinoic acid receptor or vitamin D receptor. |
|
The ligand-binding region of the receptor protein — always at the carboxyl terminus — is specific to the particular receptor. For example, in the ligand-binding region, the glucocorticoid receptor is only 30% similar to the estrogen receptor and 17% similar to the thyroid hormone receptor. The size of the ligand-binding region varies dramatically; in the vitamin D receptor it has only 25 amino acid residues, whereas in the mineralocorticoid receptor it has 603 residues. Mutations that change one amino acid residue in these regions can result in loss of responsiveness to a specific hormone. Some humans unable to respond to cortisol, testosterone, vitamin D, or thyroxine have mutations of this type.
The lncRNAs introduce another dimension to regulation by hormone receptors. An lncRNA called GAS5 (growth arrest specific 5) inhibits transcriptional activation by the glucocorticoid receptor by directly competing with DNA for receptor binding. GAS5 also inhibits activity of the closely related androgen, progesterone, and mineralocorticoid receptors. In addition, GAS5 interacts with and sequesters an miRNA called miR-21, which interacts with and inhibits the activity of some regulatory proteins that act as tumor suppressors. Expression of GAS5 is suppressed in a wide range of tumors, resulting in increased expression of steroid hormones, higher levels of active miR-21, and faster tumor growth. Low GAS5 levels thus correlate with worsened outcomes for cancer patients, making this lncRNA a subject of intense ongoing investigation.
 The lncRNAs introduce another dimension to regulation by hormone receptors. An lncRNA called GAS5 (growth arrest specific 5) inhibits transcriptional activation by the glucocorticoid receptor by directly competing with DNA for receptor binding. GAS5 also inhibits activity of the closely related androgen, progesterone, and mineralocorticoid receptors. In addition, GAS5 interacts with and sequesters an miRNA called miR-21, which interacts with and inhibits the activity of some regulatory proteins that act as tumor suppressors. Expression of GAS5 is suppressed in a wide range of tumors, resulting in increased expression of steroid hormones, higher levels of active miR-21, and faster tumor growth. Low GAS5 levels thus correlate with worsened outcomes for cancer patients, making this lncRNA a subject of intense ongoing investigation.
The lncRNAs introduce another dimension to regulation by hormone receptors. An lncRNA called GAS5 (growth arrest specific 5) inhibits transcriptional activation by the glucocorticoid receptor by directly competing with DNA for receptor binding. GAS5 also inhibits activity of the closely related androgen, progesterone, and mineralocorticoid receptors. In addition, GAS5 interacts with and sequesters an miRNA called miR-21, which interacts with and inhibits the activity of some regulatory proteins that act as tumor suppressors. Expression of GAS5 is suppressed in a wide range of tumors, resulting in increased expression of steroid hormones, higher levels of active miR-21, and faster tumor growth. Low GAS5 levels thus correlate with worsened outcomes for cancer patients, making this lncRNA a subject of intense ongoing investigation.Some hormone receptors, including the human progesterone receptor, activate transcription with the aid of a different lncRNA of ~700 nucleotides that acts as a coactivator — steroid receptor RNA activator (SRA). SRA is part of a ribonucleoprotein complex, but it is the RNA component that is required for transcription coactivation. The detailed set of interactions between SRA and other components of the regulatory systems for these genes remains to be worked out.
Regulation Can Result from Phosphorylation of Nuclear Transcription Factors
We noted in Chapter 12 that the effects of insulin on gene expression are mediated by a series of steps leading ultimately to the activation of a protein kinase in the nucleus that phosphorylates specific DNA-binding proteins, thereby altering their ability to act as transcription factors (see Fig. 12-22). This general mechanism mediates the effects of many nonsteroid hormones. For example, the β-adrenergic pathway that leads to elevated levels of cytosolic cAMP, which acts as a second messenger in both eukaryotes and bacteria (Fig. 28-18), also affects the transcription of a set of genes, each of which is located near a specific DNA sequence called a cAMP response element (CRE). The catalytic subunit of protein kinase A, released when cAMP levels rise (see Fig. 12-6), enters the nucleus and phosphorylates a nuclear protein, the CRE-binding protein (CREB). When phosphorylated, CREB binds to CREs near certain genes and acts as a transcription factor, turning on expression of these genes.
Many Eukaryotic mRNAs Are Subject to Translational Repression
Regulation at the level of translation assumes a much more prominent role in eukaryotes than in bacteria and is observed in a range of cellular situations. In contrast to the tight coupling of transcription and translation in bacteria, the transcripts generated in a eukaryotic nucleus must be processed and transported to the cytoplasm before translation. This can impose a significant delay on the appearance of a protein. When a rapid increase in protein production is needed, a translationally repressed mRNA already in the cytoplasm can be activated for translation without delay. Translational regulation may play an especially important role in regulating certain very long eukaryotic genes (a few are measured in the millions of base pairs), for which transcription and mRNA processing can require many hours. Some genes are regulated at both the transcriptional and translational stages, with the latter playing a role in the fine-tuning of cellular protein levels. In some non-nucleated cells, such as reticulocytes (immature erythrocytes), transcriptional control is entirely unavailable and translational control of stored mRNAs becomes essential. As described below, translational controls can also have spatial significance during development, when the regulated translation of prepositioned mRNAs creates a local gradient of the protein product.
 Regulation at the level of translation assumes a much more prominent role in eukaryotes than in bacteria and is observed in a range of cellular situations. In contrast to the tight coupling of transcription and translation in bacteria, the transcripts generated in a eukaryotic nucleus must be processed and transported to the cytoplasm before translation. This can impose a significant delay on the appearance of a protein. When a rapid increase in protein production is needed, a translationally repressed mRNA already in the cytoplasm can be activated for translation without delay. Translational regulation may play an especially important role in regulating certain very long eukaryotic genes (a few are measured in the millions of base pairs), for which transcription and mRNA processing can require many hours. Some genes are regulated at both the transcriptional and translational stages, with the latter playing a role in the fine-tuning of cellular protein levels. In some non-nucleated cells, such as reticulocytes (immature erythrocytes), transcriptional control is entirely unavailable and translational control of stored mRNAs becomes essential. As described below, translational controls can also have spatial significance during development, when the regulated translation of prepositioned mRNAs creates a local gradient of the protein product.
Regulation at the level of translation assumes a much more prominent role in eukaryotes than in bacteria and is observed in a range of cellular situations. In contrast to the tight coupling of transcription and translation in bacteria, the transcripts generated in a eukaryotic nucleus must be processed and transported to the cytoplasm before translation. This can impose a significant delay on the appearance of a protein. When a rapid increase in protein production is needed, a translationally repressed mRNA already in the cytoplasm can be activated for translation without delay. Translational regulation may play an especially important role in regulating certain very long eukaryotic genes (a few are measured in the millions of base pairs), for which transcription and mRNA processing can require many hours. Some genes are regulated at both the transcriptional and translational stages, with the latter playing a role in the fine-tuning of cellular protein levels. In some non-nucleated cells, such as reticulocytes (immature erythrocytes), transcriptional control is entirely unavailable and translational control of stored mRNAs becomes essential. As described below, translational controls can also have spatial significance during development, when the regulated translation of prepositioned mRNAs creates a local gradient of the protein product.Eukaryotes have at least four main mechanisms of translational regulation:
- Translation initiation factors are subject to phosphorylation by protein kinases. The phosphorylated forms are often less active and cause a general depression of translation in the cell.
- Some proteins bind directly to mRNA and act as translational repressors, many of them binding at specific sites in the untranslated region (UTR). So positioned, these proteins interact with other translation initiation factors bound to the mRNA, or with the 40S ribosomal subunit, to prevent translation initiation (Fig. 28-36).
- Binding proteins, present in eukaryotes from yeast to mammals, disrupt the interaction between eIF4E and eIF4G (see Fig. 27-27). The mammalian versions are known as 4E-BPs (eIF4E binding proteins). When cell growth is slow, these proteins limit translation by binding to the site on eIF4E that normally interacts with eIF4G. When cell growth resumes or increases in response to growth factors or other stimuli, the binding proteins are inactivated by protein kinase–dependent phosphorylation.
- RNA-mediated regulation of gene expression often occurs at the level of translational repression, often by the binding of ncRNAs to mRNAs.
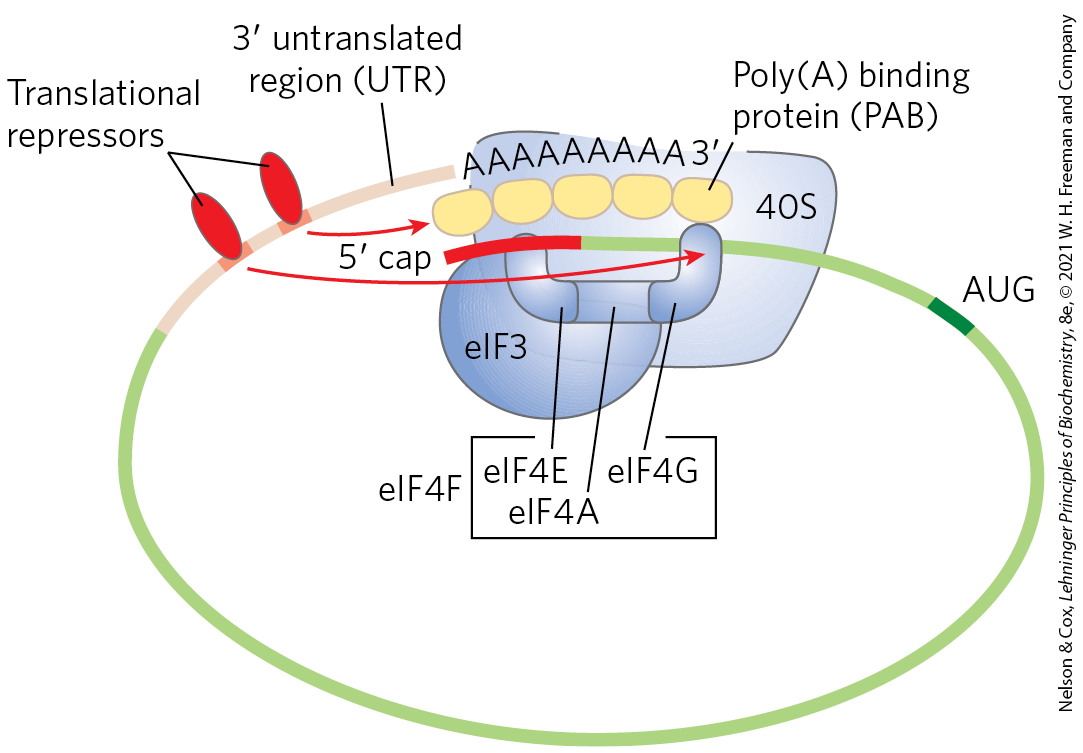
FIGURE 28-36 Translational regulation of eukaryotic mRNA. One of the most important mechanisms for translational regulation in eukaryotes is the binding of translational repressors (RNA-binding proteins) to specific sites in the untranslated region of the mRNA. These proteins interact with eukaryotic initiation factors or with the ribosome to prevent or slow translation.
The variety of translational regulation mechanisms provides flexibility, allowing focused repression of a few mRNAs or global regulation of all cellular translation.
Translational regulation has been particularly well studied in reticulocytes. One such mechanism in these cells involves eIF2, the initiation factor that binds to the initiator tRNA and conveys it to the ribosome; when Met-tRNA has bound to the P site, the factor eIF2B binds to eIF2, recycling it with the aid of GTP binding and hydrolysis. The maturation of reticulocytes includes destruction of the cell nucleus, leaving behind a plasma membrane packed with hemoglobin. Messenger RNAs deposited in the cytoplasm before the loss of the nucleus allow for the replacement of hemoglobin. When reticulocytes become deficient in iron or heme, the translation of globin mRNAs is repressed. A protein kinase called HCR (hemin-controlled repressor) is then activated, catalyzing the phosphorylation of eIF2. When phosphorylated, eIF2 forms a stable complex with eIF2B that sequesters the eIF2, making it unavailable for participation in translation. In this way, the reticulocyte coordinates the synthesis of globin with the availability of heme.
Posttranscriptional Gene Silencing Is Mediated by RNA Interference
In higher eukaryotes, including nematodes, fruit flies, plants, and mammals, microRNAs (miRNAs) mediate the silencing of many genes. In a phenomenon first described and explained by Craig Mello and Andrew Fire, the RNAs function by interacting with mRNAs, often in the , resulting in either degradation of the mRNA or inhibition of translation. In either case, the mRNA, and thus the gene that produces it, is silenced. This form of gene regulation controls developmental timing in at least some organisms. It is also used as a mechanism to protect against invading RNA viruses (particularly important in plants, which lack an immune system) and to control the activity of transposons. In addition, small RNA molecules may play a critical (as yet undefined) role in the formation of heterochromatin.
Many miRNAs are present only transiently during development, and these are sometimes referred to as small temporal RNAs (stRNAs). Thousands of different miRNAs have been identified in higher eukaryotes, and they may affect the regulation of a third of mammalian genes. They are transcribed as precursor RNAs ~70 nucleotides long, with internally complementary sequences that form hairpinlike structures. Details of the pathway for processing of miRNAs were described in Fig. 26-26). The precursors are cleaved by endonucleases such as Drosha and Dicer to form short duplexes of 20 to 25 nucleotides. One strand of the processed miRNA is transferred to the target mRNA (or to a viral or transposon RNA), leading to inhibition of translation or degradation of the mRNA (Fig. 28-37a). Some miRNAs bind to and affect a single mRNA and thus affect expression of only one gene. Others interact with multiple mRNAs and form the mechanistic core of regulons that coordinate the expression of multiple genes.
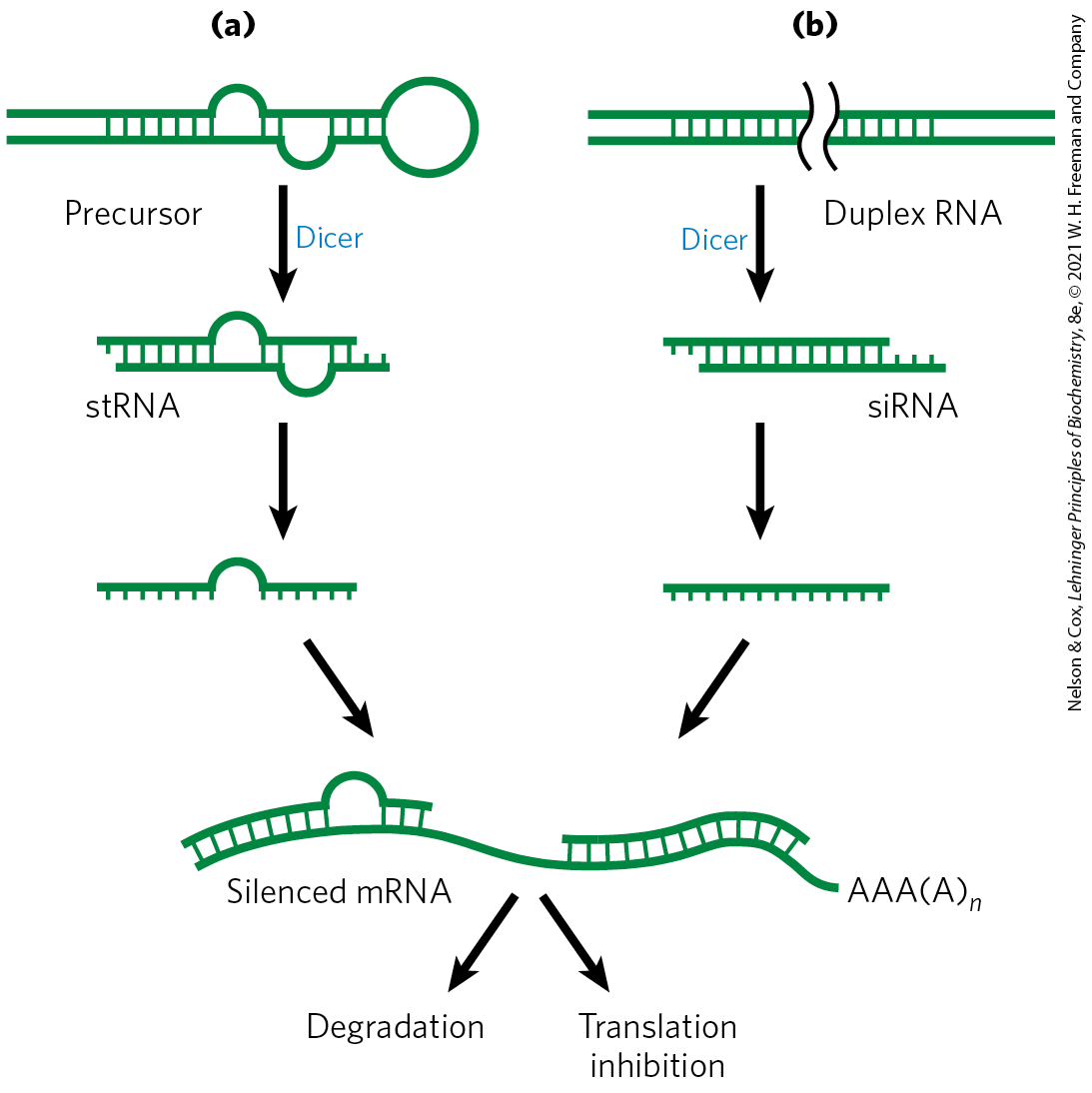
FIGURE 28-37 Gene silencing by RNA interference. (a) Small temporal RNAs (stRNAs, a class of miRNAs) are generated by Dicer-mediated cleavage of longer precursors that fold to create duplex regions. The stRNAs then bind to mRNAs, leading to degradation of mRNA or inhibition of translation. (b) Double-stranded RNAs designed to interact with a particular target and to function as Dicer substrates can be constructed and introduced into a cell. Dicer processes the duplex RNAs into small interfering RNAs (siRNAs), which interact with the target mRNA. Again, either the mRNA is degraded or translation is inhibited.
This gene regulation mechanism has an interesting and very useful practical side. If an investigator introduces into an organism a duplex RNA molecule corresponding in sequence to virtually any mRNA, Dicer cleaves the duplex into short segments, called small interfering RNAs (siRNAs). These bind to the mRNA and silence it (Fig. 28-37b). The process is known as RNA interference (RNAi). In plants, almost any gene can be effectively shut down in this way. Nematodes can readily ingest entire functional RNAs, and simply introducing the duplex RNA into the worm’s diet produces very effective suppression of the target gene. The technique is an important tool in the ongoing efforts to study gene function, because it can disrupt gene function without creating a mutant organism. The procedure can be applied to humans as well. Laboratory-produced siRNAs have been used to block HIV and poliovirus infections in cultured human cells for a week or so at a time. The wider application of RNAi-based pharmaceuticals was initially stymied by the difficulty inherent in delivering RNAi molecules to their required target, given the many nucleases that degrade RNA in human tissues. With recent advances in delivery methods, there are now more than a dozen RNAi pharmaceuticals in advanced clinical trials to treat a range of conditions, from familial amyloidotic polyneuropathy to viral infections and cancer.
RNA-Mediated Regulation of Gene Expression Takes Many Forms in Eukaryotes
All RNAs (regardless of their length) that do not encode proteins, including rRNAs and tRNAs, come under the general designation of ncRNAs. Mammalian genomes encode more ncRNAs than coding mRNAs. The ncRNAs in eukaryotes include miRNAs, described above; snRNAs, involved in RNA splicing (see Fig. 26-16); snoRNAs, involved in rRNA modification (see Fig. 26-24); and lncRNAs, already encountered in this chapter. Not surprisingly, additional functional classes of ncRNAs are still being discovered. Here we describe a few more examples of ncRNAs that participate in gene regulation, which are designated lncRNAs when their length exceeds 200 nucleotides.
Heat shock factor 1 (HSF1) is an activator protein that, in nonstressed cells, exists as a monomer bound by the chaperone Hsp90. Under stress conditions, HSF1 is released from Hsp90 and trimerizes. The HSF1 trimer binds to DNA and activates transcription of genes encoding products required to deal with the stress. An lncRNA called HSR1 (heat shock RNA 1; ∼600 nucleotides) stimulates HSF1 trimerization and DNA binding. HSR1 does not act alone; it functions in a complex with the translation elongation factor eEF1A.
Additional RNAs affect transcription in a variety of ways. A 331 nucleotide lncRNA called 7SK, abundant in mammals, binds to the Pol II transcription elongation factor pTEFb (see Table 26-2) and represses transcript elongation. The ncRNA B2 (∼178 nucleotides) binds directly to Pol II during heat shock and represses transcription. The B2-bound Pol II assembles into stable PICs, but transcription is blocked. The mechanism that allows HSF1-responsive genes to be expressed in the presence of B2 remains to be worked out.
The recognized roles of ncRNAs in gene expression and in many other cellular processes are rapidly expanding. At the same time, the study of the biochemistry of gene regulation is becoming much less protein-centric.
Development Is Controlled by Cascades of Regulatory Proteins
For sheer complexity and intricacy of coordination, the patterns of gene regulation that bring about development of a zygote into a multicellular animal or plant have no peer. Development requires transitions in morphology and protein composition that depend on tightly coordinated changes in expression of the genome. More genes are expressed during early development than in any other part of the life cycle. For example, in the sea urchin, an oocyte has about 18,500 different mRNAs, compared with about 6,000 different mRNAs in the cells of a typical differentiated tissue. The mRNAs in the oocyte give rise to a cascade of events that regulate the expression of many genes across both space and time.
Several organisms have emerged as important model systems for the study of development, because they are easy to maintain in a laboratory and have relatively short generation times. These include nematodes, fruit flies, zebra fish, mice, and the plant Arabidopsis. Here, we provide a brief discussion of the development of fruit flies. Our understanding of the molecular events during development of Drosophila melanogaster is particularly well advanced and can be used to illustrate patterns and principles of general significance.
The life cycle of the fruit fly includes complete metamorphosis during its progression from an embryo to an adult (Fig. 28-38). Among the most important characteristics of the embryo are its polarity (the anterior and posterior parts of the animal are readily distinguished, as are its dorsal and ventral surfaces) and its metamerism (the embryo body is made up of serially repeating segments, each with characteristic features). During development, these segments become organized into a head, thorax, and abdomen. Each segment of the adult thorax has a different set of appendages. Development of this complex pattern is under genetic control, and a variety of pattern-regulating genes have been discovered that greatly affect the organization of the body.
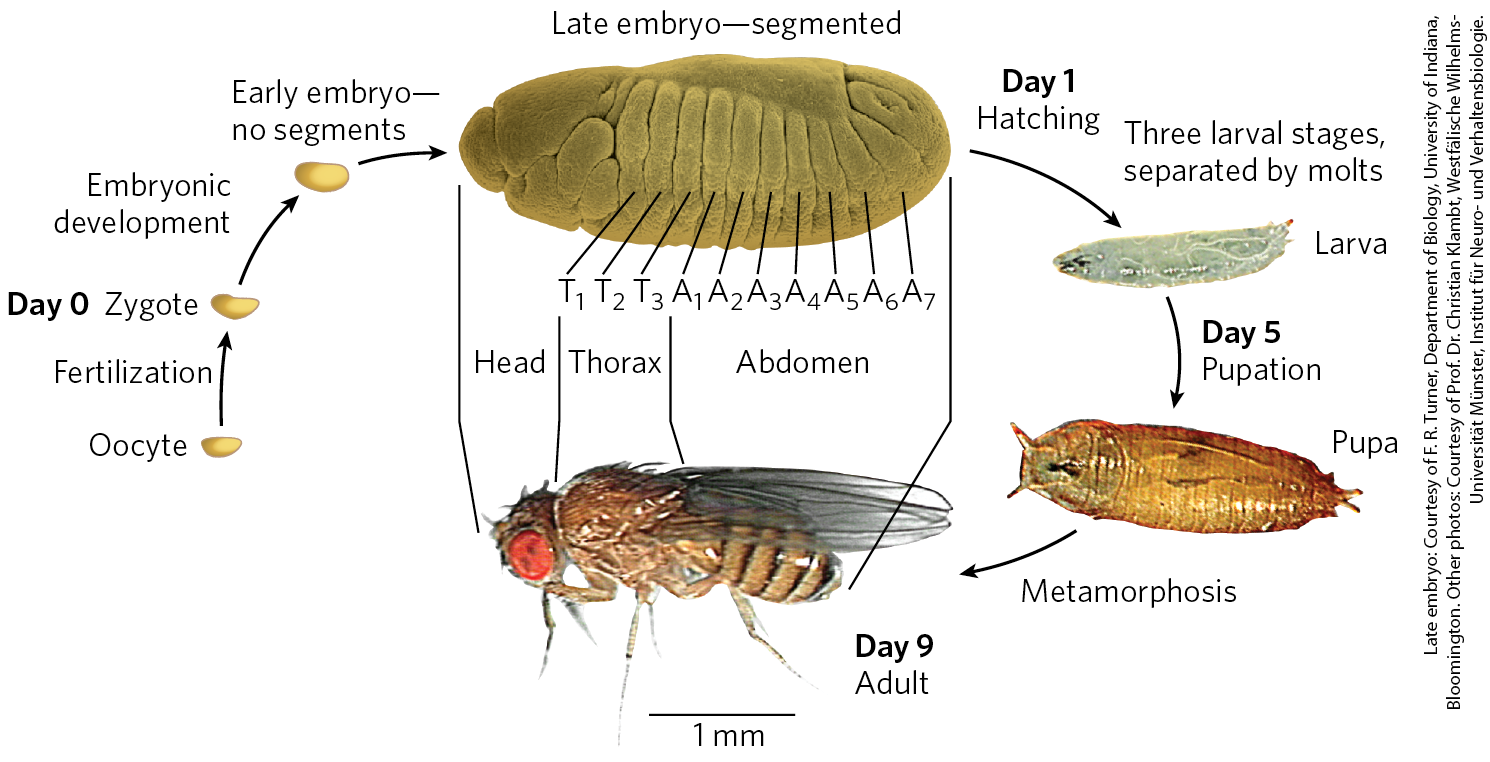
FIGURE 28-38 Life cycle of the fruit fly Drosophila melanogaster. Drosophila undergoes a complete metamorphosis, which means that the adult insect is radically different in form from its immature stages, a transformation that requires extensive alterations during development. By the late embryonic stage, segments have formed, each containing specialized structures from which the various appendages and other features of the adult fly will develop.
The Drosophila egg, along with 15 nurse cells, is surrounded by a layer of follicle cells (Fig. 28-39). As the egg cell forms (before fertilization), mRNAs and proteins originating in the nurse and follicle cells are deposited in the egg cell, where some play a critical role in development. Once a fertilized egg is laid, its nucleus divides and the nuclear descendants continue to divide in synchrony every 6 to 10 min. Plasma membranes are not formed around the nuclei, which are distributed within the egg cytoplasm, forming a syncytium. Between the eighth and eleventh rounds of nuclear division, the nuclei migrate to the outer layer of the egg, forming a monolayer of nuclei surrounding the common yolk-rich cytoplasm; this is the syncytial blastoderm. After a few additional divisions, membrane invaginations surround the nuclei to create a layer of cells that form the cellular blastoderm. At this stage, the mitotic cycles in the various cells lose their synchrony. The developmental fate of the cells is determined by the mRNAs and proteins originally deposited in the egg by the nurse and follicle cells.
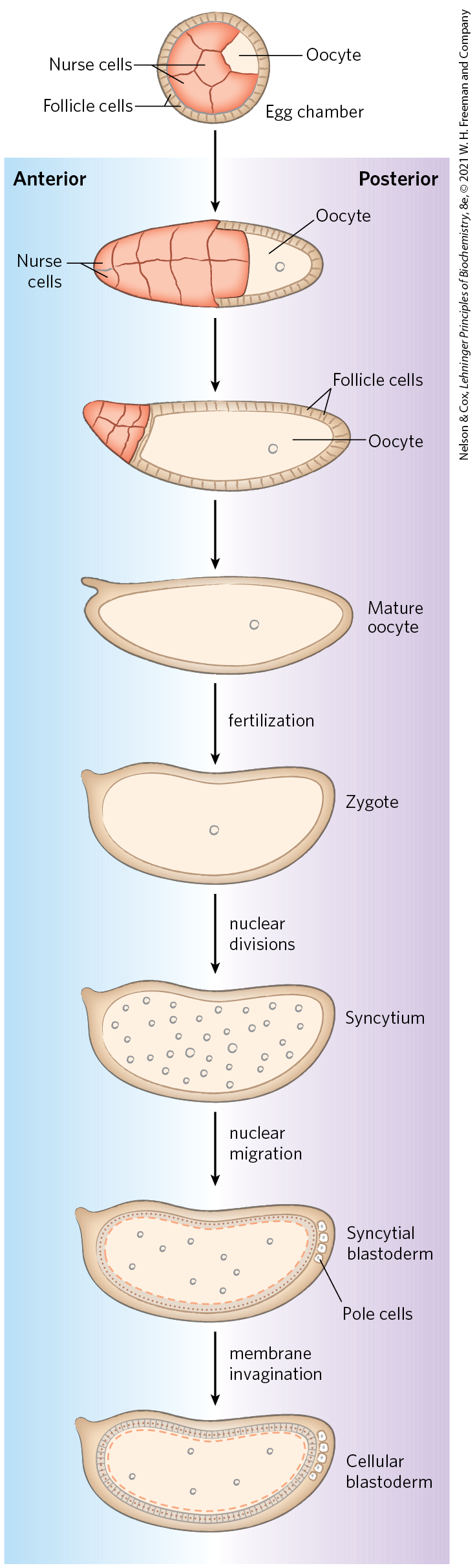
FIGURE 28-39 Early development in Drosophila. During development of the egg, maternal mRNAs and proteins are deposited in the developing oocyte (unfertilized egg cell) by nurse cells and follicle cells. After fertilization, the nuclei of the egg divide in synchrony within the common cytoplasm (syncytium), then migrate to the periphery. Membrane invaginations surround the nuclei to create a monolayer of cells at the periphery; this is the cellular blastoderm stage. During the early nuclear divisions, several nuclei at the far posterior become pole cells, which later become the germ-line cells.
Proteins that, through changes in local concentration or activity, cause the surrounding tissue to take up a particular shape or structure are sometimes referred to as morphogens; they are the products of pattern-regulating genes. As defined by Christiane Nüsslein-Volhard, Edward B. Lewis, and Eric F. Wieschaus, three major classes of pattern-regulating genes — maternal, segmentation, and homeotic genes — function in successive stages of development to specify the basic features of the Drosophila embryo body. Maternal genes are expressed in the unfertilized egg, and the resulting maternal mRNAs remain dormant until fertilization. These provide most of the proteins needed in very early development, until the cellular blastoderm is formed. Some of the proteins encoded by maternal mRNAs direct the spatial organization of the developing embryo at early stages, establishing its polarity. Segmentation genes, transcribed after fertilization, direct the formation of the proper number of body segments. At least three subclasses of segmentation genes act at successive stages: gap genes divide the developing embryo into several broad regions; pair-rule genes, together with segment polarity genes, define 14 stripes that become the 14 segments of a normal embryo. Homeotic genes are expressed still later; they specify which organs and appendages will develop in particular body segments.
If all cells divided to produce two identical daughter cells, multicellular organisms would never be more than a ball of identical cells. A key event in very early development is establishment of mRNA and protein gradients along the body axes, producing asymmetric cell divisions and different cell fates. Some maternal mRNAs have protein products that diffuse through the cytoplasm to create an asymmetric distribution in the egg. Different cells in the cellular blastoderm therefore inherit different amounts of these proteins, setting the cells on different developmental paths. An example is the bicoid gene. The bicoid gene product is a major anterior morphogen. The mRNA from the bicoid gene is synthesized by nurse cells and deposited in the unfertilized egg near its anterior pole. Translated soon after fertilization, the Bicoid protein diffuses through the cell to create, by the seventh nuclear division, a concentration gradient radiating out from the anterior pole (Fig. 28-40). The Bicoid protein contains a homeodomain (p. 1062), encoded by a gene sequence motif called a homeobox and found in many proteins involved in regulating development. Bicoid is multifunctional — a transcription factor that activates the expression of several segmentation genes and also a translational repressor that inactivates certain mRNAs. The amount of Bicoid protein in various parts of the embryo increases or decreases the expression of other genes in a threshold-dependent manner. As its concentration varies along its gradient, interactions of the bicoid gene product with proteins and RNAs encoded by the nanos, pumilio, caudal, hunchback, and other regulatory genes also vary to produce different effects along the axis of the developing organism. This results in different developmental fates of cells in the blastoderm, depending on their location.
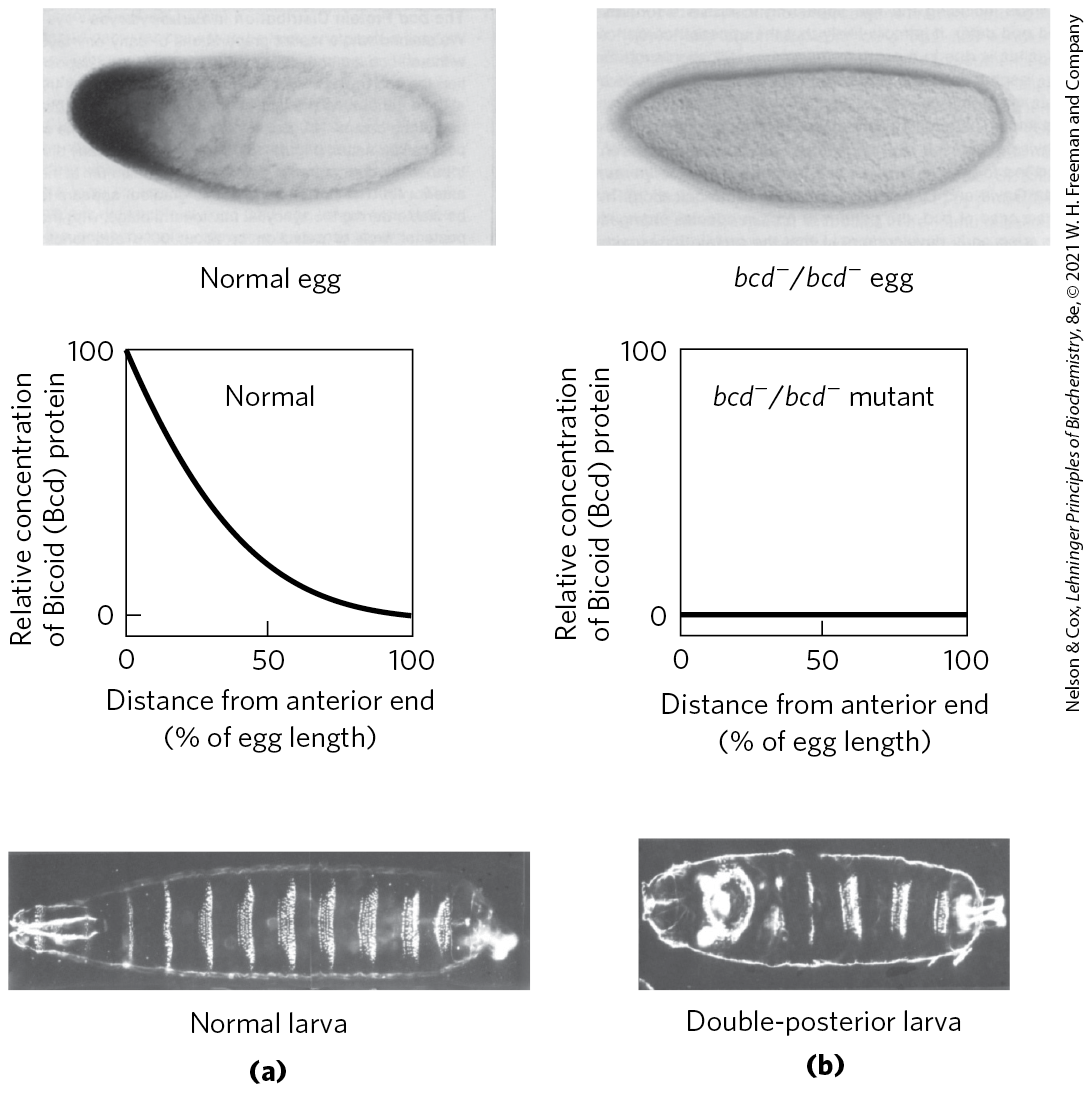
FIGURE 28-40 Distribution of a maternal gene product in a Drosophila egg. (a) Micrograph of an immunologically stained egg (top), showing distribution of the bicoid (bcd) gene product. The graph shows stain intensity along the length of the egg. This distribution is essential for normal development of the anterior structures in the larva (bottom). (b) If the bcd gene is not expressed by the mother and thus no bicoid mRNA is deposited in the egg, the resulting larva has two posteriors (and soon dies). [Republished with permission of Elsevier, from “The bicoid protein determines position in the Drosophila embryo in a concentration-dependent manner” by Wolfgang Driever and Christiane Nüsslein-Volhard, Cell 54:83–93, July 1, 1988; permission conveyed through Copyright Clearance Center, Inc.]
Humans do not resemble fruit flies, but the genes and mechanisms involved in development are nevertheless highly conserved. This can be seen in the gene clusters encoding the homeotic or Hox genes, the latter term derived from homeobox. Drosophila has one such cluster, while humans have four (Fig. 28-41), with the genes within the clusters remarkably similar from nematodes to humans.
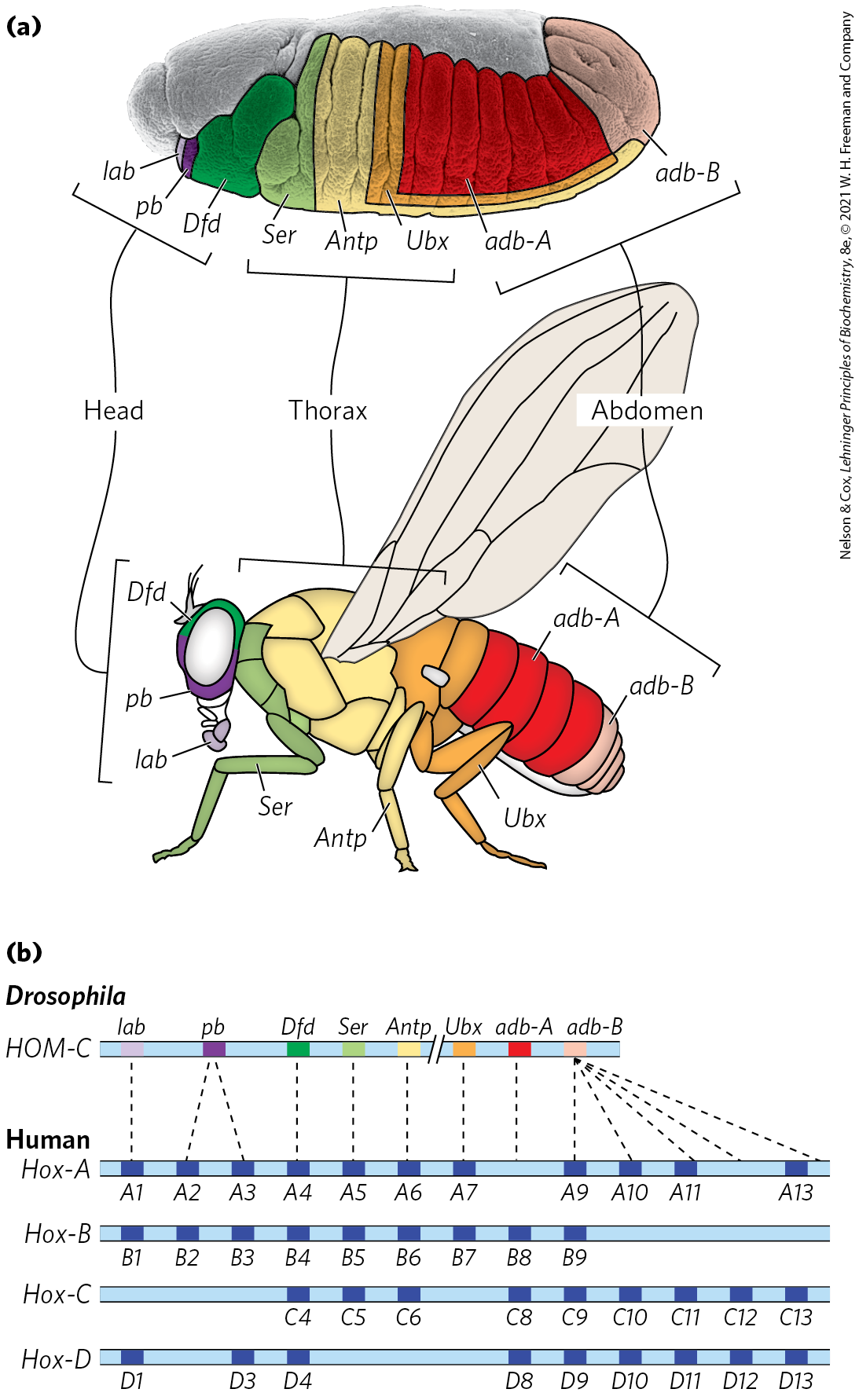
FIGURE 28-41 The Hox gene clusters and their effects on development. (a) Each Hox gene in the fruit fly is responsible for the development of structures in a defined part of the body and is expressed in defined regions of the embryo, as labeled. (b) Drosophila has one Hox gene cluster; the human genome has four. Many of these genes are highly conserved in multicellular animals. Evolutionary relationships, as indicated by sequence alignments, between genes in the fruit fly Hox gene cluster and those in the mammalian Hox gene clusters are shown by dashed lines. Similar relationships among the four sets of mammalian Hox genes are indicated by vertical alignment. [(a) Information from F. R. Turner, University of Indiana, Department of Biology.]
The many regulatory genes in these three classes direct the development of an adult fly, with a head, thorax, and abdomen, with the proper number of segments, and with the correct appendages on each segment. Although embryogenesis takes about a day to complete, all these genes are activated during the first four hours. Some mRNAs and proteins are present for only a few minutes at specific points during this period. Some of the genes code for transcription factors that affect the expression of other genes in a kind of developmental cascade. Regulation at the level of translation also occurs, and many of the regulatory genes encode translational repressors, most of which bind to the UTR of the mRNA (Fig. 28-36). Because many mRNAs are deposited in the egg long before their translation is required, translational repression provides an especially important avenue for regulation in developmental pathways.
Many of the principles of development outlined above apply to other eukaryotes, from nematodes to humans. Some of the regulatory proteins are conserved. For example, the products of the homeobox-containing genes HOXA7 in mouse and antennapedia in fruit fly differ in only one amino acid residue. Of course, although the molecular regulatory mechanisms may be similar, many of the ultimate developmental events are not conserved (humans do not have wings or antennae). The different outcomes are brought about by differences in the downstream target genes controlled by the Hox genes. The discovery of structural determinants with identifiable molecular functions is the first step in understanding the molecular events underlying development. As more genes and their protein products are discovered, the biochemical side of this vast puzzle will be elucidated in increasingly rich detail.
Stem Cells Have Developmental Potential That Can Be Controlled
If we can understand development, and the mechanisms of gene regulation behind it, we can control it. An adult human has many different types of tissues. Many of the cells are terminally differentiated and no longer divide. If an organ malfunctions due to disease, or a limb is lost in an accident, the tissues are not readily replaced. Most cells, because of the regulatory processes in place, or even because of the loss of some or all of the genomic DNA, are not easily reprogrammed. Medical science has made organ transplants possible, but organ donors are a limited resource and organ rejection remains a major medical problem. If humans could regenerate their own organs or limbs or nervous tissue, rejection would no longer be an issue. Cures for kidney failure or neurodegenerative disorders could become reality.
The key to tissue regeneration lies in stem cells — cells that have retained the capacity to differentiate into various tissues. In humans, after an egg is fertilized, the first few cell divisions create a ball of totipotent cells, called the morula, that have the capacity to differentiate individually into any tissue or even into a complete organism (Fig. 28-42). Continued cell division produces a hollow ball, the blastocyst. The outer cells of the blastocyst eventually form the placenta. The inner layers form the germ layers of the developing fetus — the ectoderm, mesoderm, and endoderm. These cells are pluripotent: they can give rise to cells of all three germ layers and can differentiate into many types of tissues. However, they cannot differentiate into a complete organism. Some of these cells are unipotent: they can develop into only one type of cell and/or tissue. It is the pluripotent cells of the blastocyst, the embryonic stem cells, that are currently used in embryonic stem cell research.
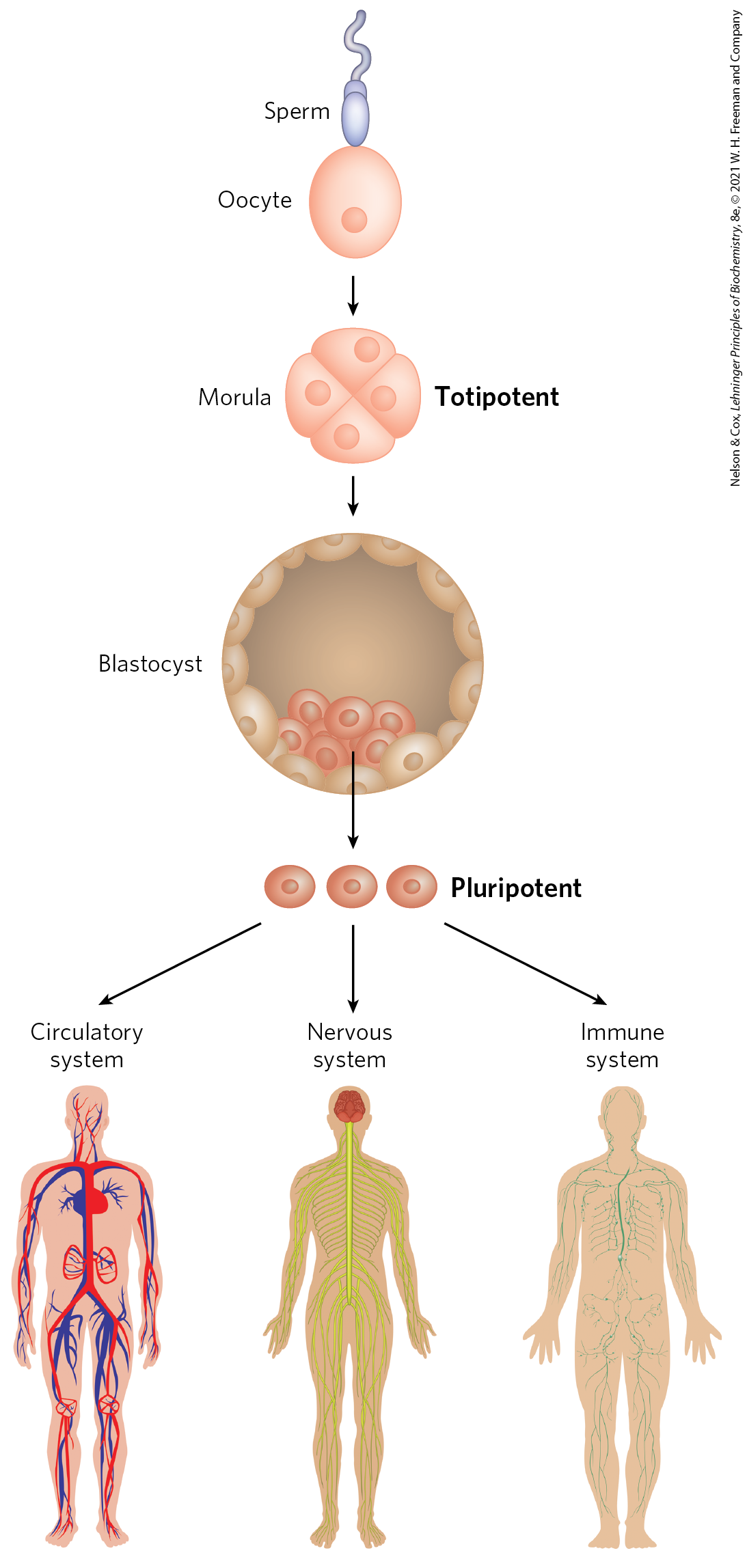
FIGURE 28-42 Totipotent and pluripotent stem cells. Cells at the morula stage are totipotent and have the capacity to differentiate into a complete organism. The source of pluripotent embryonic stem cells is the cells in the cavity of the blastocyst. Pluripotent cells give rise to many tissue types but cannot form complete organisms.
Stem cells have two functions: to replenish themselves and, at the same time, provide cells that can differentiate. These tasks are accomplished in multiple ways (Fig. 28-43a). All or parts of the stem cell population can, in principle, be involved in replenishment, differentiation, or both.
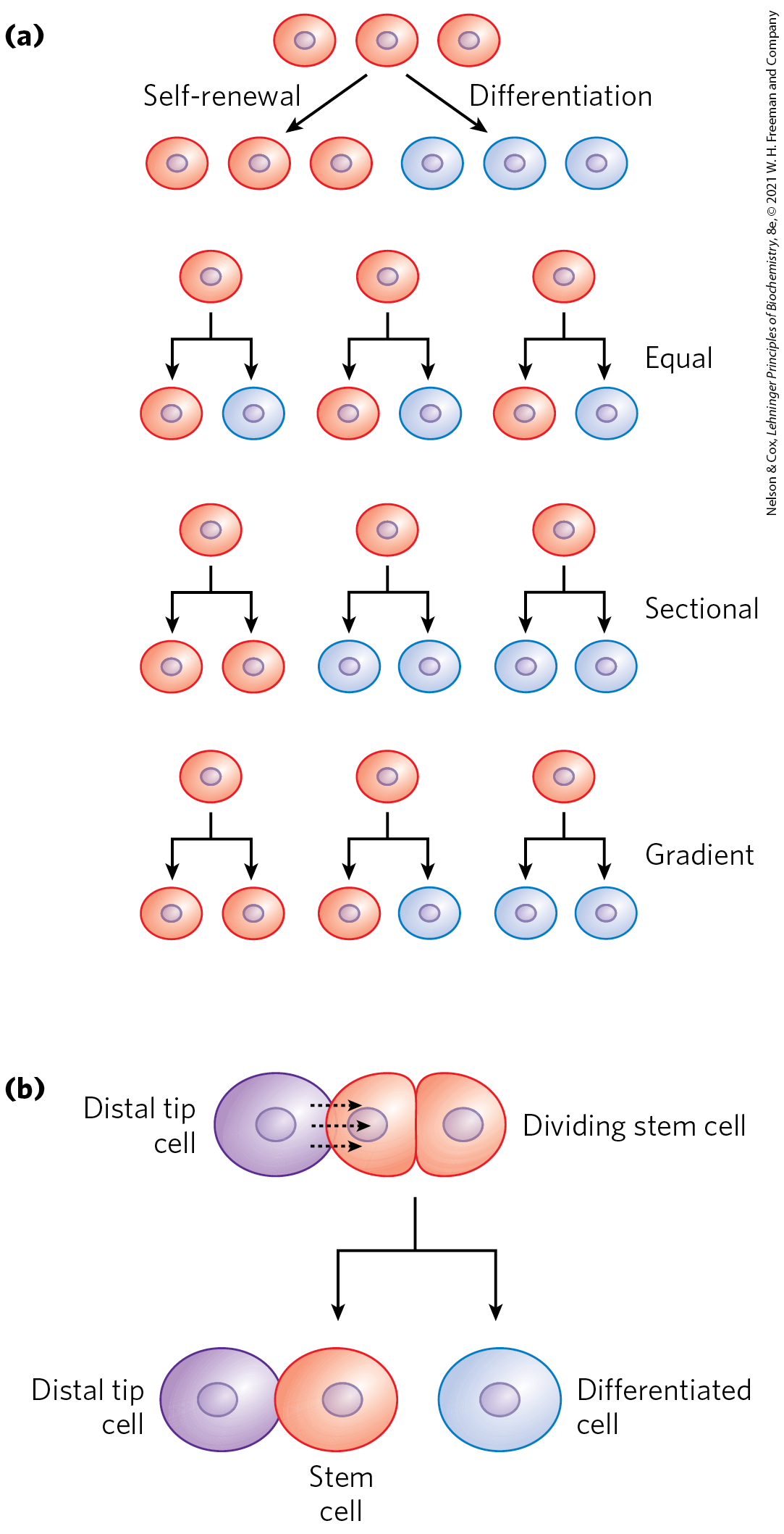
FIGURE 28-43 Stem cell proliferation versus differentiation and development. Stem cells must strike a balance between self-renewal and differentiation. (a) Some possible cell division patterns that allow the replenishment of stem cells and production of some differentiated cells. Each cell may produce one stem cell and one differentiated cell, or two differentiated cells, or two stem cells in defined parts of the tissue or culture. Or a gradient of growth conditions can be established, with cell fates differing from one end of the gradient to the other. (b) Establishing a developmental niche through stem cell contact with a cell or group of cells. Molecular signals provided by the niche cells (in this case, in plants, a distal tip cell) help orient the mitotic spindle for stem cell division and ensure that one daughter cell retains stem cell properties.
Other types of stem cells can potentially be used for medical benefit. In the adult organism, adult stem cells, as products of additional differentiation, have a more limited potential for further development than do embryonic stem cells. For example, the hematopoietic stem cells of bone marrow can give rise to many types of blood cells and also to cells with the capacity to regenerate bone. They are referred to as multipotent. However, these cells cannot differentiate into a liver or kidney or neuron. Adult stem cells are often said to have a niche, a microenvironment that promotes stem cell maintenance while allowing differentiation of some daughter cells as replacements for cells in the tissue they serve (Fig. 28-43b). Hematopoietic stem cells in the bone marrow occupy a niche in which signaling from neighboring cells and other cues maintain the stem cell lineage. At the same time, some daughter cells differentiate to provide needed blood cells. Understanding the niche in which stem cells operate, and the signals the niche provides, is essential in efforts to harness the potential of stem cells for tissue regeneration. The identification and culturing of pluripotent stem cells from human blastocysts was reported by James Thomson and colleagues in 1998. This advance led to the long-term availability of established cell lines for research.
All stem cells present problems for human medical applications. Adult stem cells have a limited capacity to regenerate tissues, are generally present in small numbers, and are hard to isolate from an adult human. Embryonic stem cells have much greater differentiation potential and can be cultured to generate large numbers of cells, but their use is accompanied by ethical concerns related to the necessary destruction of human embryos. Identifying a source of plentiful and medically useful stem cells that does not raise such concerns remains a major goal of medical research.
Our ability to culture stem cells (i.e., maintain them in an undifferentiated state), and to manipulate them to grow and differentiate into particular tissues, is very much a function of our understanding of developmental biology.
Thus far, mouse and human embryonic stem cells have been used for most research. Although both types of stem cells are pluripotent, they require very different culture conditions, optimized to allow cell division indefinitely without differentiation. Mouse embryonic stem cells are grown on a layer of gelatin and require the presence of leukemia inhibitory factor (LIF). Human embryonic stem cells are grown on a feeder layer of mouse embryonic fibroblasts and require basic fibroblast growth factor (bFGF, or FGF2). The use of a feeder cell layer implies that the mouse cells are providing a diffusible product or some surface signal, not yet known, that is needed by human stem cells to either promote cell division or prevent differentiation.
A significant advance, reported in 2007, centers on success in reversing differentiation. In effect, skin cells — first from mice, then from humans — have been reprogrammed to take on the characteristics of pluripotent stem cells. The reprogramming involves manipulations to get the cells to express at least four transcription factors, Oct4, Sox2, Nanog, and Lin28, all of which are known to help maintain the stem cell–like state. Gradual improvements in this technology may make the harvesting of embryonic stem cells unnecessary and provide a source of stem cells that is genetically matched to a prospective patient.
Our discussion of developmental regulation and stem cells brings us full circle, back to a biochemical beginning. Evolution appropriately provides the first and last words of this book. If evolution is to generate the kind of changes in an organism that would render it a different species, it is the developmental program that must be affected. Developmental and evolutionary processes are closely allied, each informing the other (Box 28-1). The continuing study of biochemistry has everything to do with enriching the future of humanity and understanding our origins.
SUMMARY 28.3 Regulation of Gene Expression in Eukaryotes
- In eukaryotes, large changes in chromatin structure accompany the expression of a gene. Transcriptionally inactive heterochromatin is opened up by chromatin remodeling proteins. These eject, replace, or modify nucleosomes to allow other proteins, mainly RNA polymerase components and regulators, to access sites required to initiate transcription.
- In eukaryotes, positive regulation is more common than negative regulation.
- Promoters for Pol II typically have a TATA box and Inr sequence, as well as multiple binding sites for transcription activators. The latter sites, sometimes located hundreds or thousands of base pairs away from the TATA box, are called upstream activator sequences in yeast and enhancers in higher eukaryotes. To regulate transcriptional activity generally requires large complexes of proteins. These include basal transcription factors, activators, coactivators, architectural regulators, and the enzymes that modify and remodel chromatin. The effects of transcription activators on Pol II are facilitated by coactivator protein complexes such as Mediator.
- The well-studied yeast genes involved in galactose metabolism provide examples of both positive and negative regulation in a eukaryote.
- The modular structures of the activators have distinct activation and DNA-binding domains.
- Hormones affect the regulation of gene expression in one of two ways. Steroid hormones interact directly with intracellular receptors that are DNA-binding regulatory proteins; binding of the hormone has either positive or negative effects on the transcription of targeted genes.
- Nonsteroid hormones bind to cell surface receptors, triggering a signaling pathway that can lead to phosphorylation of a regulatory protein, affecting its activity.
- Translational regulation is particularly important in eukaryotes. Modulating the translation of an mRNA stored in the cytoplasm affords a more rapid response to cellular challenges than de novo assembly of transcription complexes and mRNA synthesis.
- MicroRNAs (miRNAs) are involved in gene silencing during development and as an antiviral defense. The pathway for processing miRNAs from larger precursors has been harnessed by researchers to develop the gene-silencing technology called RNA interference, or RNAi.
- Regulation mediated by ncRNAs plays an important role in eukaryotic gene expression, with known mechanisms including interactions with proteins, mRNA, and other ncRNAs.
- Development of a multicellular organism presents the most complex regulatory challenge. The fate of cells in the early embryo is determined by establishment of anterior-posterior and dorsal-ventral gradients of proteins that act as transcription activators or translational repressors, regulating the genes required for development of structures appropriate to a particular part of the organism. Sets of regulatory genes operate in temporal and spatial succession, transforming given areas of an egg cell into predictable structures in the adult organism.
- The differentiation of stem cells into functional tissues can be controlled by extracellular signals and conditions.
 In eukaryotes, large changes in chromatin structure accompany the expression of a gene. Transcriptionally inactive heterochromatin is opened up by chromatin remodeling proteins. These eject, replace, or modify nucleosomes to allow other proteins, mainly RNA polymerase components and regulators, to access sites required to initiate transcription.
In eukaryotes, large changes in chromatin structure accompany the expression of a gene. Transcriptionally inactive heterochromatin is opened up by chromatin remodeling proteins. These eject, replace, or modify nucleosomes to allow other proteins, mainly RNA polymerase components and regulators, to access sites required to initiate transcription.