In ureotelic organisms, the ammonia deposited in the mitochondria of hepatocytes is converted to urea in the urea cycle (Fig. 18-10). This pathway was discovered in 1932 by Hans Krebs (who later also discovered the citric acid cycle) and a medical student associate, Kurt Henseleit. Urea production occurs almost exclusively in the liver and is the fate of most of the ammonia channeled there. The urea passes into the bloodstream and thus to the kidneys and is excreted into the urine. The production of urea now becomes the focus of our discussion.
FIGURE 18-10 The urea cycle and reactions that feed amino groups into the cycle. The enzymes catalyzing these reactions (named in the text) are distributed between the mitochondrial matrix and the cytosol. One amino group enters the urea cycle as carbamoyl phosphate, formed in the matrix; the other enters as aspartate, formed in the matrix by transamination of oxaloacetate and glutamate, catalyzed by aspartate aminotransferase. The urea cycle consists of four steps: Formation of citrulline from ornithine and carbamoyl phosphate (entry of the first amino group); the citrulline passes into the cytosol. Formation of argininosuccinate through a citrullyl-AMP intermediate (entry of the second amino group). Formation of arginine from argininosuccinate; this reaction releases fumarate, which enters the citric acid cycle. Formation of urea; this reaction also regenerates ornithine.
Urea Is Produced from Ammonia in Five Enzymatic Steps
The urea cycle begins inside liver mitochondria, but three of the subsequent steps take place in the cytosol; the cycle thus spans two cellular compartments (Fig. 18-10). The first amino group to enter the urea cycle is derived from ammonia in the mitochondrial matrix — most of this arises by the pathways described in Section 18.1. The liver also receives some ammonia via the portal vein from the intestine, from the bacterial oxidation of amino acids. Whatever its source, the generated in liver mitochondria is immediately used, together with (as ) produced by mitochondrial respiration, to form carbamoyl phosphate in the matrix (as shown in Fig. 18-10 and explained in detail in Fig. 18-11a). This ATP-dependent reaction is catalyzed by carbamoyl phosphate synthetase I, a regulatory enzyme (see below). The mitochondrial form of the enzyme is distinct from the cytosolic (II) form, which has a separate function in pyrimidine biosynthesis (Chapter 22).
MECHANISM FIGURE 18-11 Nitrogen-acquiring reactions in the synthesis of urea. The urea nitrogens are acquired in two separate reactions, each requiring ATP. (a) In the first reaction, catalyzed by carbamoyl phosphate synthetase I, the first nitrogen enters from ammonia. The terminal phosphate groups of two molecules of ATP are used to form one molecule of carbamoyl phosphate. In other words, this reaction has two activation steps ( and ). (b) In the second reaction, catalyzed by argininosuccinate synthetase, the second nitrogen enters from aspartate. This reaction has two steps. Activation of the ureido oxygen of citrulline in step sets up the addition of aspartate in step .
The carbamoyl phosphate, which functions as an activated carbamoyl group donor, now enters the urea cycle. The cycle has only four enzymatic steps. First, carbamoyl phosphate donates its carbamoyl group to ornithine to form citrulline, with the release of (Fig. 18-10, step ). The reaction is catalyzed by ornithine transcarbamoylase. Ornithine is not one of the 20 common amino acids found in proteins, but it is a key intermediate in arginine biosynthesis and nitrogen metabolism in general. It is synthesized from glutamate in a five-step pathway described in Chapter 22. Ornithine plays a role resembling that of oxaloacetate in the citric acid cycle, accepting material at each turn of the urea cycle. The citrulline produced in the first step of the urea cycle passes from the mitochondrion to the cytosol.
The next two steps bring in the second amino group, featuring aspartate as the amino group donor. The aspartate is generated in the mitochondria by transamination between glutamate and oxaloacetate, and then transported into the cytosol. A condensation reaction between the amino group of aspartate and the ureido (carbonyl) group of citrulline forms argininosuccinate (step in Fig. 18-10). This cytosolic reaction, catalyzed by argininosuccinate synthetase, requires ATP and proceeds through a citrullyl-AMP intermediate (Fig. 18-11b). The argininosuccinate is then cleaved by argininosuccinase (step in Fig. 18-10) to form free arginine and fumarate, the latter being converted to malate before entering mitochondria to join the pool of citric acid cycle intermediates. This is the only reversible step in the urea cycle. In the last reaction of the urea cycle (step ), the cytosolic enzyme arginase cleaves arginine to yield urea and ornithine. Ornithine is transported into the mitochondrion to initiate another round of the urea cycle.
As we noted in Chapter 16, the enzymes of many metabolic pathways are clustered in metabolons (p. 595), with the product of one enzyme reaction being channeled directly to the next enzyme in the pathway. In the urea cycle, the mitochondrial and cytosolic enzymes seem to be clustered in this way. The citrulline transported out of the mitochondrion is not diluted into the general pool of metabolites in the cytosol but is passed directly to the active site of argininosuccinate synthetase. This channeling between enzymes continues for argininosuccinate, arginine, and ornithine. Only urea is released into the general cytosolic pool of metabolites.
The Citric Acid and Urea Cycles Can Be Linked
The fumarate produced in the argininosuccinase reaction is also an intermediate of the citric acid cycle. Thus, the cycles are, in principle, interconnected — in a process dubbed the “Krebs bicycle” (Fig. 18-12). However, each cycle can operate independently, and communication between them depends on the transport of key intermediates between the mitochondrion and cytosol. Major transporters in the inner mitochondrial membrane include the malate–α-ketoglutarate transporter, the glutamate-aspartate transporter, and the glutamate- transporter. Together, these transporters facilitate the movement of malate and glutamate into the mitochondrial matrix and the movement of aspartate and α-ketoglutarate out to the cytosol.
FIGURE 18-12 Links between the urea cycle and citric acid cycle. The interconnected cycles have been called the “Krebs bicycle.” The pathways linking the citric acid and urea cycles are known as the aspartate-argininosuccinate shunt; these effectively link the fates of the amino groups and the carbon skeletons of amino acids. The interconnections are quite elaborate. For example, some citric acid cycle enzymes, such as fumarase and malate dehydrogenase, have both cytosolic and mitochondrial isozymes. Fumarate produced in the cytosol — whether by the urea cycle, purine biosynthesis, or other processes — can be converted to cytosolic malate, which is used in the cytosol or transported into mitochondria to enter the citric acid cycle. These processes are further intertwined with the malate-aspartate shuttle, a set of reactions that brings reducing equivalents into the mitochondrion. These different cycles and processes rely on a limited number of transporters in the inner mitochondrial membrane.
Several enzymes of the citric acid cycle, including fumarase (fumarate hydratase) and malate dehydrogenase (p. 586), are also present as isozymes in the cytosol. There is no transporter to directly move the fumarate generated in cytosolic arginine synthesis back into the mitochondrial matrix. However, fumarate can be converted to malate in the cytosol. Fumarate and malate can be further metabolized in the cytosol, or malate can be transported into mitochondria for use in the citric acid cycle. Aspartate formed in mitochondria by transamination between oxaloacetate and glutamate can be transported to the cytosol, where it serves as nitrogen donor in the urea cycle reaction catalyzed by argininosuccinate synthetase. These reactions, making up the aspartate-argininosuccinate shunt, provide metabolic links between the separate pathways by which the amino groups and carbon skeletons of amino acids are processed.
The use of aspartate as a nitrogen donor in the urea cycle may appear to be a relatively complicated way to introduce the second amino group into urea. However, we shall see in Chapter 22 that this pathway for nitrogen incorporation is one of the two common ways to introduce amino groups into biomolecules. In the urea cycle, additional pathway interconnections can help explain why aspartate is used as a nitrogen donor. The urea and citric acid cycles are closely tied to an additional process that brings NADH, in the form of reducing equivalents, into the mitochondrion. As detailed in the next chapter, the NADH produced by glycolysis, fatty acid oxidation, and other processes cannot be transported across the mitochondrial inner membrane. Reducing equivalents are instead brought into the mitochondrion by converting aspartate to oxaloacetate in the cytosol, reducing the oxaloacetate to malate with NADH, and transporting the malate into the mitochondrial matrix via the malate–α-ketoglutarate transporter. Once inside the mitochondrion, the malate can be reconverted to oxaloacetate while generating NADH. The oxaloacetate is converted to aspartate in the matrix and transported out of the mitochondrion by the aspartate-glutamate transporter. This malate-aspartate shuttle completes yet another cycle that functions to keep the mitochondrion supplied with NADH (Fig. 18-12; see also Fig. 19-31).
These processes require that a balance be maintained in the cytosol between the concentrations of glutamate and aspartate. The enzyme that transfers amino groups between these key amino acids is aspartate aminotransferase, AAT (also called glutamate-oxaloacetate transaminase, GOT). This is one of the most active enzymes in hepatocytes and other tissues. When tissue damage occurs, this easily assayed enzyme and others leak into the blood. Thus, measuring blood levels of liver enzymes is important in diagnosing a variety of medical conditions (Box 18-1).
The Activity of the Urea Cycle Is Regulated at Two Levels
The flux of nitrogen through the urea cycle in an individual animal varies with diet. When the dietary intake is primarily protein, the carbon skeletons of amino acids are used for fuel, producing much urea from the excess amino groups. During prolonged starvation, when breakdown of muscle protein begins to supply much of the organism’s metabolic energy, urea production also increases substantially.
These changes in demand for urea cycle activity are met over the long term by regulation of the rates of synthesis of the four urea cycle enzymes and carbamoyl phosphate synthetase I in the liver. All five enzymes are synthesized at higher rates in starving animals and in animals on very-high-protein diets than in well-fed animals eating primarily carbohydrates and fats. Animals on protein-free diets produce lower levels of urea cycle enzymes.
On a shorter time scale, allosteric regulation of at least one key enzyme adjusts the flux through the urea cycle. The first enzyme in the pathway, carbamoyl phosphate synthetase I, is allosterically activated by N-acetylglutamate, which is synthesized from acetyl-CoA and glutamate by N-acetylglutamate synthase (Fig. 18-13). In plants and microorganisms, this enzyme catalyzes the first step in the de novo synthesis of arginine from glutamate (see Fig. 22-12), but in mammals, N-acetylglutamate synthase activity in the liver has a purely regulatory function (mammals lack the other enzymes needed to convert glutamate to arginine). The steady-state levels of N-acetylglutamate are determined by the concentrations of glutamate and acetyl-CoA (the substrates for N-acetylglutamate synthase) and arginine (an activator of N-acetylglutamate synthase, and thus an activator of the urea cycle).
FIGURE 18-13 Synthesis of N-acetylglutamate and its activation of carbamoyl phosphate synthetase I.
Pathway Interconnections Reduce the Energetic Cost of Urea Synthesis
If we consider the urea cycle in isolation, we see that the synthesis of one molecule of urea requires four high-energy phosphate groups (Fig. 18-10). Two ATP molecules are required to make carbamoyl phosphate, and one ATP to make argininosuccinate — the latter ATP undergoing a pyrophosphate cleavage to AMP and , which is hydrolyzed to two . The overall equation of the urea cycle is
However, this apparent cost is compensated for by the pathway interconnections detailed above. The fumarate generated by the urea cycle is converted to malate, and the malate is transported into the mitochondrion (Fig. 18-12). Inside the mitochondrial matrix, NADH is generated in the malate dehydrogenase reaction. Each NADH molecule can generate up to 2.5 ATP during mitochondrial respiration, greatly reducing the overall energetic cost of urea synthesis (mitochondrial respiration is discussed further in Chapter 19).
Genetic Defects in the Urea Cycle Can Be Life-Threatening
Infants with severe genetic defects in any enzyme involved in urea formation often appear normal at birth. However, they soon develop symptoms of hyperammonemia, including cerebral edema, lethargy, and hyperventilation. Without treatment, early death usually results. Symptoms may be less severe in patients retaining partial enzyme activity. These patients cannot tolerate protein-rich diets. Amino acids ingested in excess of the minimum daily requirements for protein synthesis are deaminated in the liver, producing free ammonia that cannot be converted to urea and exported into the bloodstream, and, as we have seen, ammonia is highly toxic. Given that most urea cycle steps are irreversible, the absent enzyme activity can often be identified by determining which cycle intermediate is present in elevated concentration in the blood and/or urine. Although the breakdown of amino acids can have serious health consequences in individuals with urea cycle deficiencies, a protein-free diet is not a treatment option. Humans are incapable of synthesizing half of the 20 common amino acids; they must obtain these essential amino acids in the diet (Table 18-1).
TABLE 18-1 Nonessential and Essential Amino Acids for Humans and the Albino Rat
aRequired to some degree in young, growing animals and/or sometimes during illness.
A variety of treatments are available for individuals with urea cycle defects. Careful administration of the aromatic acids benzoate or phenylbutyrate in the diet can help lower the level of ammonia in the blood. Benzoate is converted to benzoyl-CoA, which combines with glycine to form hippurate (Fig. 18-14, left). The glycine used up in this reaction must be regenerated, and ammonia is thus taken up in the glycine synthase reaction. Phenylbutyrate is converted to phenylacetate by β oxidation. The phenylacetate is then converted to phenylacetyl-CoA, which combines with glutamine to form phenylacetylglutamine (Fig. 18-14, right). The resulting removal of glutamine triggers its further synthesis by glutamine synthetase (see Eqn 22-1) in a reaction that takes up ammonia. Both hippurate and phenylacetylglutamine are nontoxic compounds that are excreted in the urine. The pathways shown in Figure 18-14 make only minor contributions to normal metabolism, but they become prominent when aromatic acids are ingested.
FIGURE 18-14 Treatment for deficiencies in urea cycle enzymes. The aromatic acids benzoate and phenylbutyrate, administered in the diet, are metabolized and combine with glycine and glutamine, respectively. The products are excreted in the urine. Subsequent synthesis of glycine and glutamine to replenish the pool of these intermediates removes ammonia from the bloodstream.
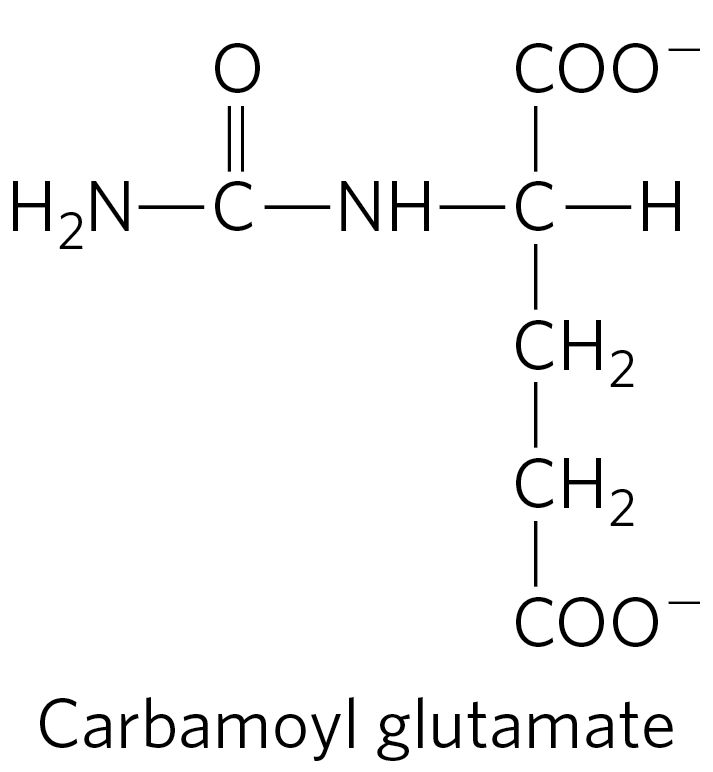
Other therapies are more specific to a particular enzyme deficiency. Deficiency of N-acetylglutamate synthase results in the absence of the normal activator of carbamoyl phosphate synthetase I (Fig. 18-13). This condition can be treated by administering carbamoyl glutamate, an analog of N-acetylglutamate that is effective in activating carbamoyl phosphate synthetase I.
Supplementing the diet with arginine is useful in treating deficiencies of ornithine transcarbamoylase, argininosuccinate synthetase, and argininosuccinase. Many of these treatments must be accompanied by strict dietary control and supplements of essential amino acids. In the rare cases of arginase deficiency, arginine, the substrate of the defective enzyme, must be excluded from the diet.
SUMMARY 18.2 Nitrogen Excretion and the Urea Cycle
In the urea cycle, ornithine combines with ammonia, in the form of carbamoyl phosphate, to form citrulline. A second amino group is transferred to citrulline from aspartate to form arginine — the immediate precursor of urea. Arginase catalyzes hydrolysis of arginine to urea and ornithine; ornithine is regenerated in each turn of the cycle.
The urea cycle results in a net conversion of oxaloacetate to fumarate, both of which are intermediates in the citric acid cycle. The two cycles are thus interconnected.
The activity of the urea cycle is regulated at the level of enzyme synthesis and by allosteric regulation of the enzyme that catalyzes the formation of carbamoyl phosphate.
The energetic cost of the urea cycle is reduced by cycle interconnections.
Genetic diseases involving urea cycle enzyme deficiencies have serious consequences but sometimes can be managed by dietary intervention.
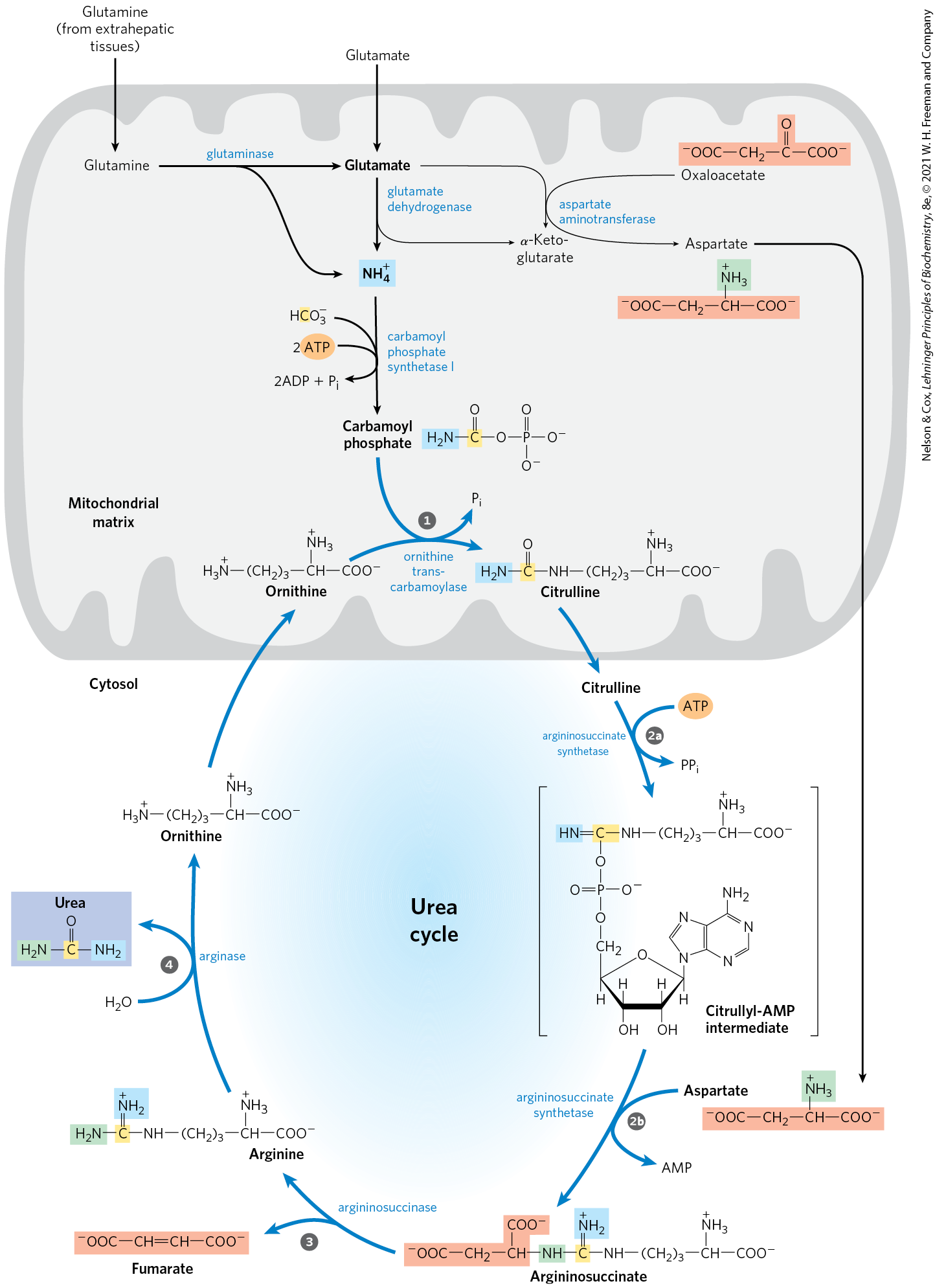
 Formation of citrulline from ornithine and carbamoyl phosphate (entry of the first amino group); the citrulline passes into the cytosol.
Formation of citrulline from ornithine and carbamoyl phosphate (entry of the first amino group); the citrulline passes into the cytosol.  Formation of argininosuccinate through a citrullyl-AMP intermediate (entry of the second amino group).
Formation of argininosuccinate through a citrullyl-AMP intermediate (entry of the second amino group).  Formation of arginine from argininosuccinate; this reaction releases fumarate, which enters the citric acid cycle.
Formation of arginine from argininosuccinate; this reaction releases fumarate, which enters the citric acid cycle.  Formation of urea; this reaction also regenerates ornithine.
Formation of urea; this reaction also regenerates ornithine.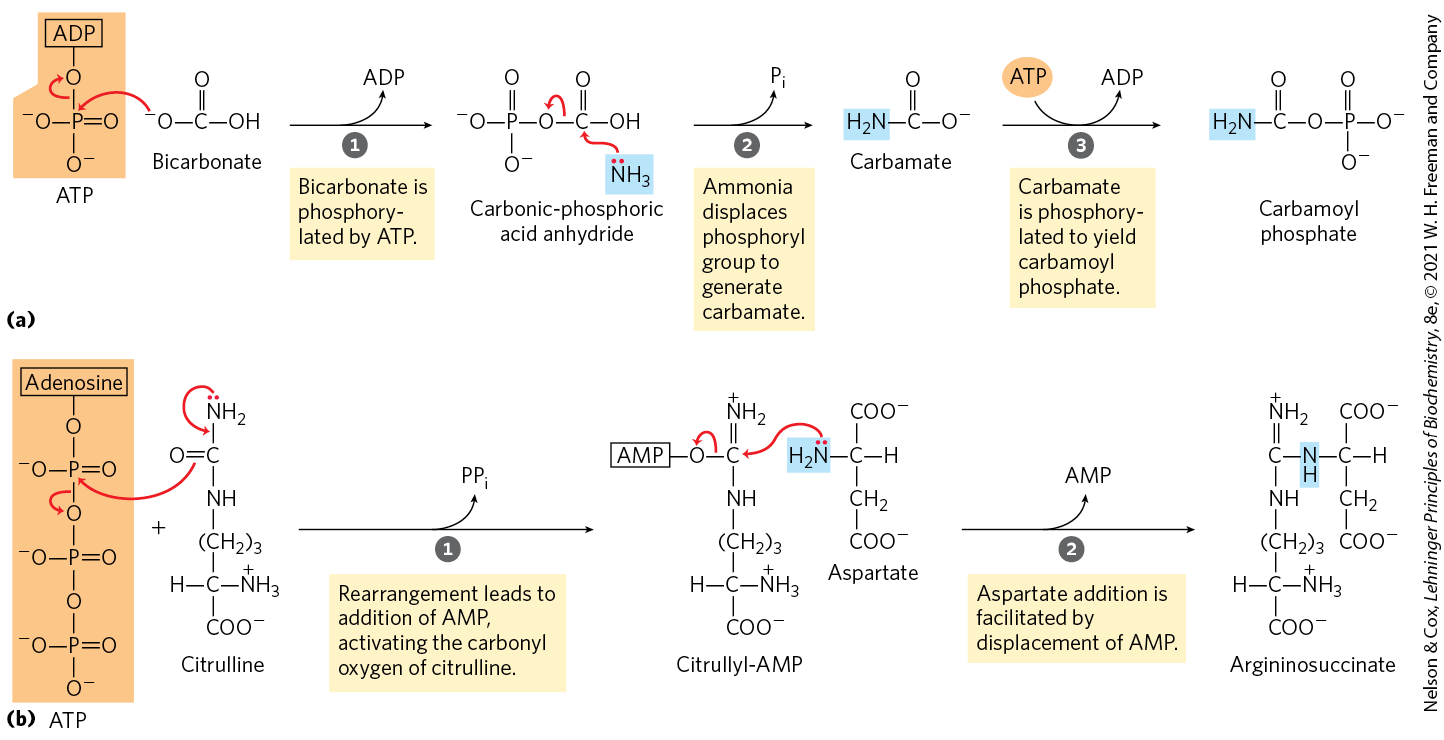
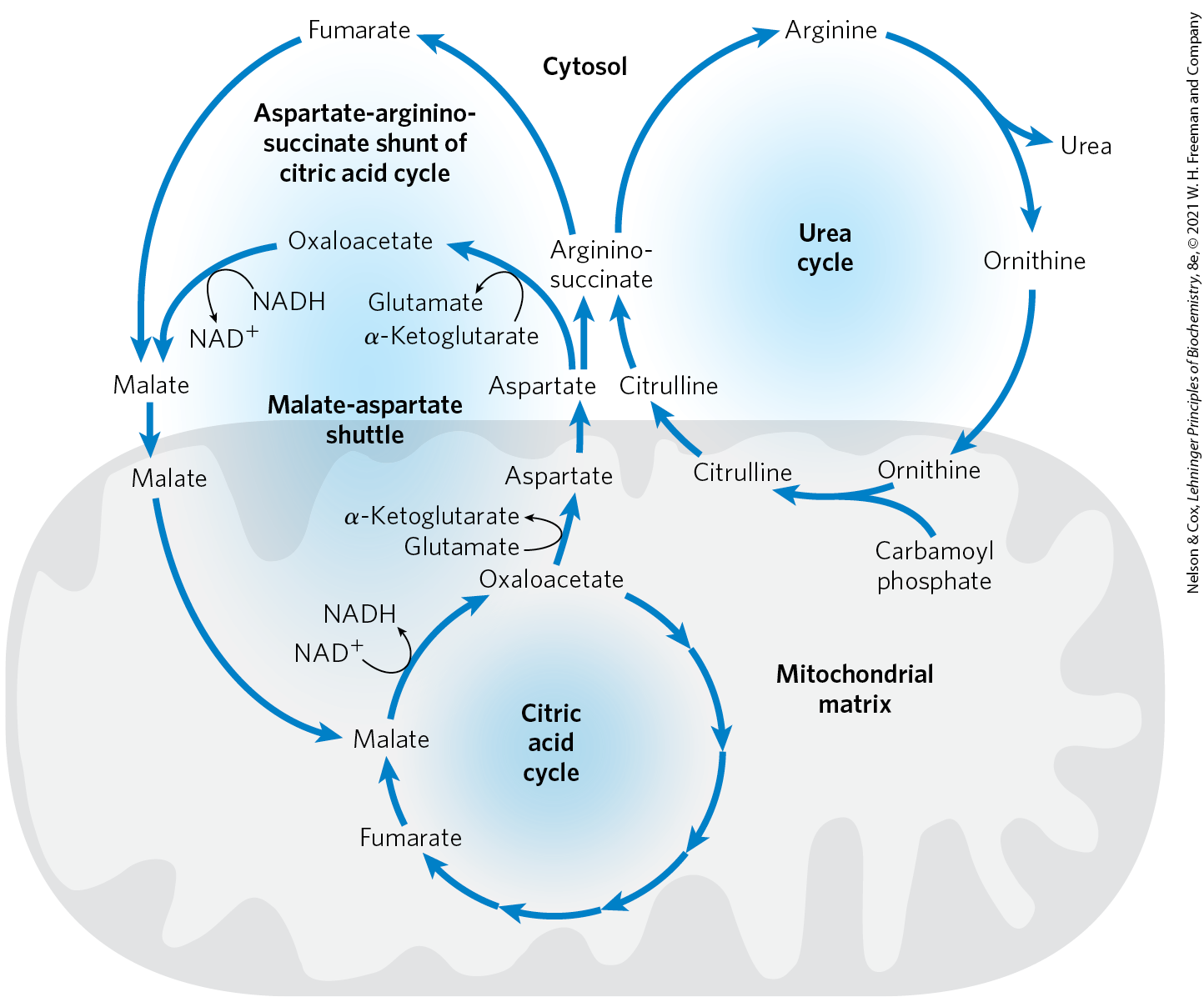
 The urea and citric acid cycles are closely tied to an additional process that brings NADH, in the form of reducing equivalents, into the mitochondrion. As detailed in the next chapter, the NADH produced by glycolysis, fatty acid oxidation, and other processes cannot be transported across the mitochondrial inner membrane. Reducing equivalents are instead brought into the mitochondrion by converting aspartate to oxaloacetate in the cytosol, reducing the oxaloacetate to malate with NADH, and transporting the malate into the mitochondrial matrix via the malate–α-ketoglutarate transporter. Once inside the mitochondrion, the malate can be reconverted to oxaloacetate while generating NADH. The oxaloacetate is converted to aspartate in the matrix and transported out of the mitochondrion by the aspartate-glutamate transporter. This malate-aspartate shuttle completes yet another cycle that functions to keep the mitochondrion supplied with NADH (
The urea and citric acid cycles are closely tied to an additional process that brings NADH, in the form of reducing equivalents, into the mitochondrion. As detailed in the next chapter, the NADH produced by glycolysis, fatty acid oxidation, and other processes cannot be transported across the mitochondrial inner membrane. Reducing equivalents are instead brought into the mitochondrion by converting aspartate to oxaloacetate in the cytosol, reducing the oxaloacetate to malate with NADH, and transporting the malate into the mitochondrial matrix via the malate–α-ketoglutarate transporter. Once inside the mitochondrion, the malate can be reconverted to oxaloacetate while generating NADH. The oxaloacetate is converted to aspartate in the matrix and transported out of the mitochondrion by the aspartate-glutamate transporter. This malate-aspartate shuttle completes yet another cycle that functions to keep the mitochondrion supplied with NADH (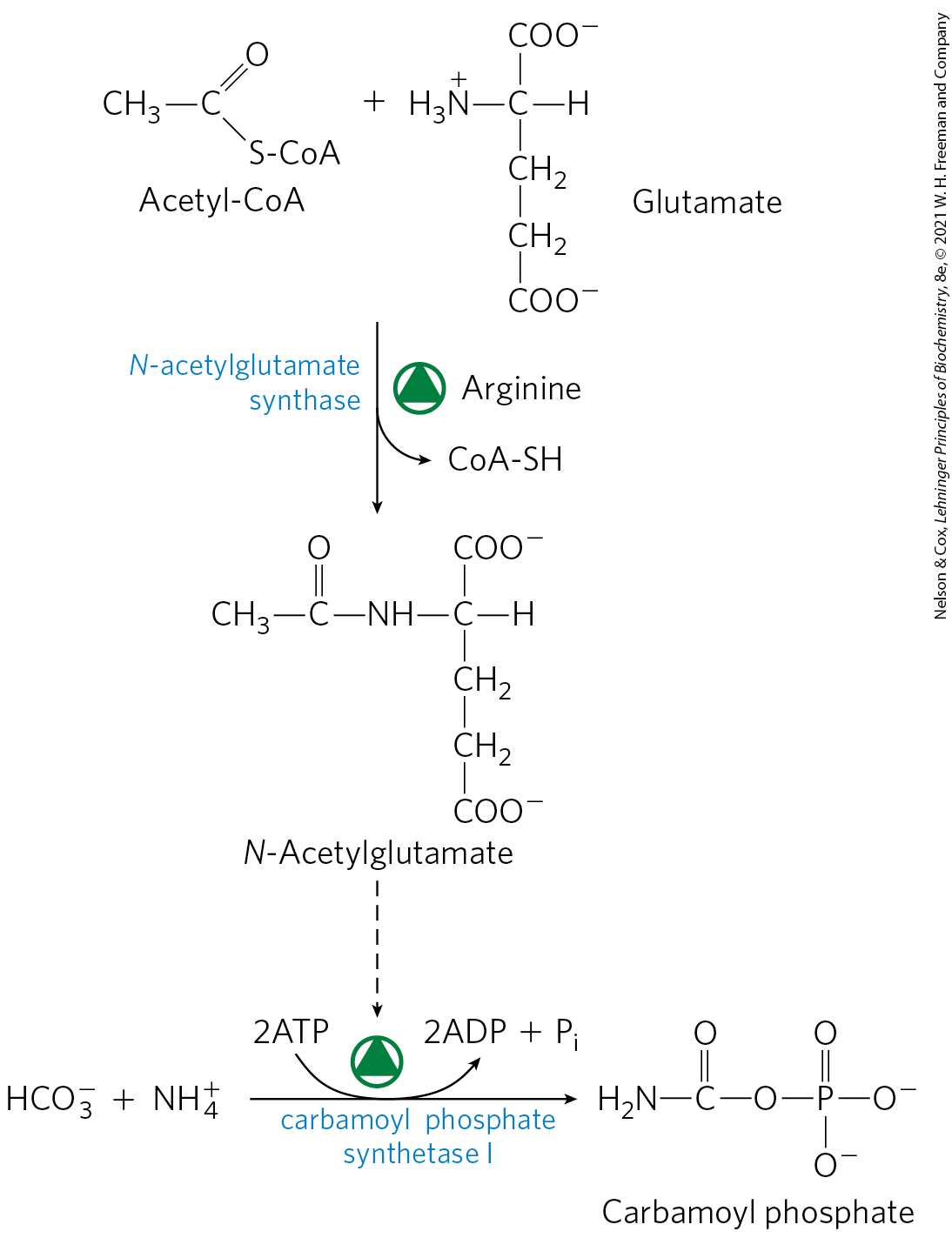
 Infants with severe genetic defects in any enzyme involved in urea formation often appear normal at birth. However, they soon develop symptoms of hyperammonemia, including cerebral edema, lethargy, and hyperventilation. Without treatment, early death usually results. Symptoms may be less severe in patients retaining partial enzyme activity. These patients cannot tolerate protein-rich diets. Amino acids ingested in excess of the minimum daily requirements for protein synthesis are deaminated in the liver, producing free ammonia that cannot be converted to urea and exported into the bloodstream, and, as we have seen, ammonia is highly toxic. Given that most urea cycle steps are irreversible, the absent enzyme activity can often be identified by determining which cycle intermediate is present in elevated concentration in the blood and/or urine. Although the breakdown of amino acids can have serious health consequences in individuals with urea cycle deficiencies, a protein-free diet is not a treatment option. Humans are incapable of synthesizing half of the 20 common amino acids; they must obtain these
Infants with severe genetic defects in any enzyme involved in urea formation often appear normal at birth. However, they soon develop symptoms of hyperammonemia, including cerebral edema, lethargy, and hyperventilation. Without treatment, early death usually results. Symptoms may be less severe in patients retaining partial enzyme activity. These patients cannot tolerate protein-rich diets. Amino acids ingested in excess of the minimum daily requirements for protein synthesis are deaminated in the liver, producing free ammonia that cannot be converted to urea and exported into the bloodstream, and, as we have seen, ammonia is highly toxic. Given that most urea cycle steps are irreversible, the absent enzyme activity can often be identified by determining which cycle intermediate is present in elevated concentration in the blood and/or urine. Although the breakdown of amino acids can have serious health consequences in individuals with urea cycle deficiencies, a protein-free diet is not a treatment option. Humans are incapable of synthesizing half of the 20 common amino acids; they must obtain these 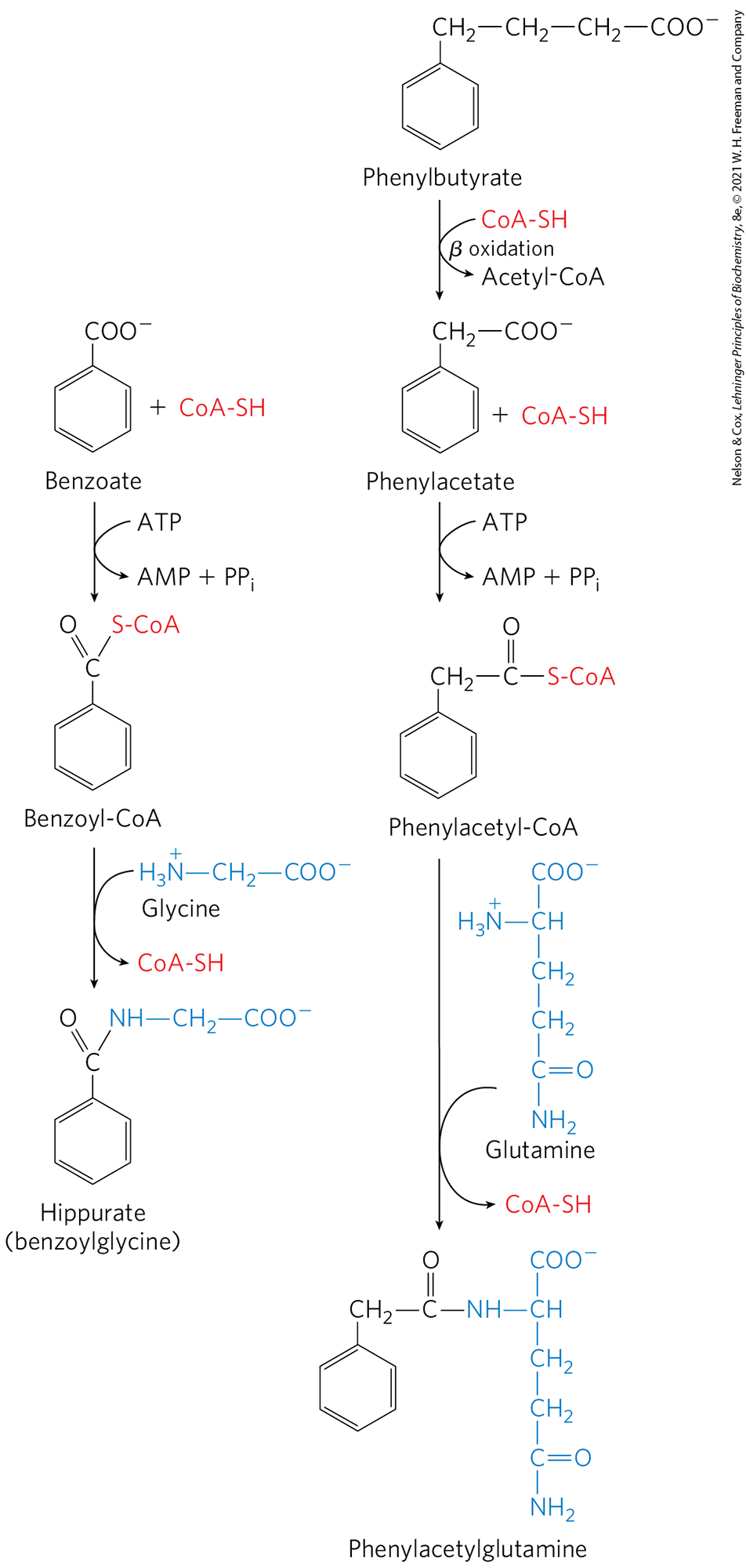

 In the urea cycle, ornithine combines with ammonia, in the form of carbamoyl phosphate, to form citrulline. A second amino group is transferred to citrulline from aspartate to form arginine — the immediate precursor of urea. Arginase catalyzes hydrolysis of arginine to urea and ornithine; ornithine is regenerated in each turn of the cycle.
In the urea cycle, ornithine combines with ammonia, in the form of carbamoyl phosphate, to form citrulline. A second amino group is transferred to citrulline from aspartate to form arginine — the immediate precursor of urea. Arginase catalyzes hydrolysis of arginine to urea and ornithine; ornithine is regenerated in each turn of the cycle.