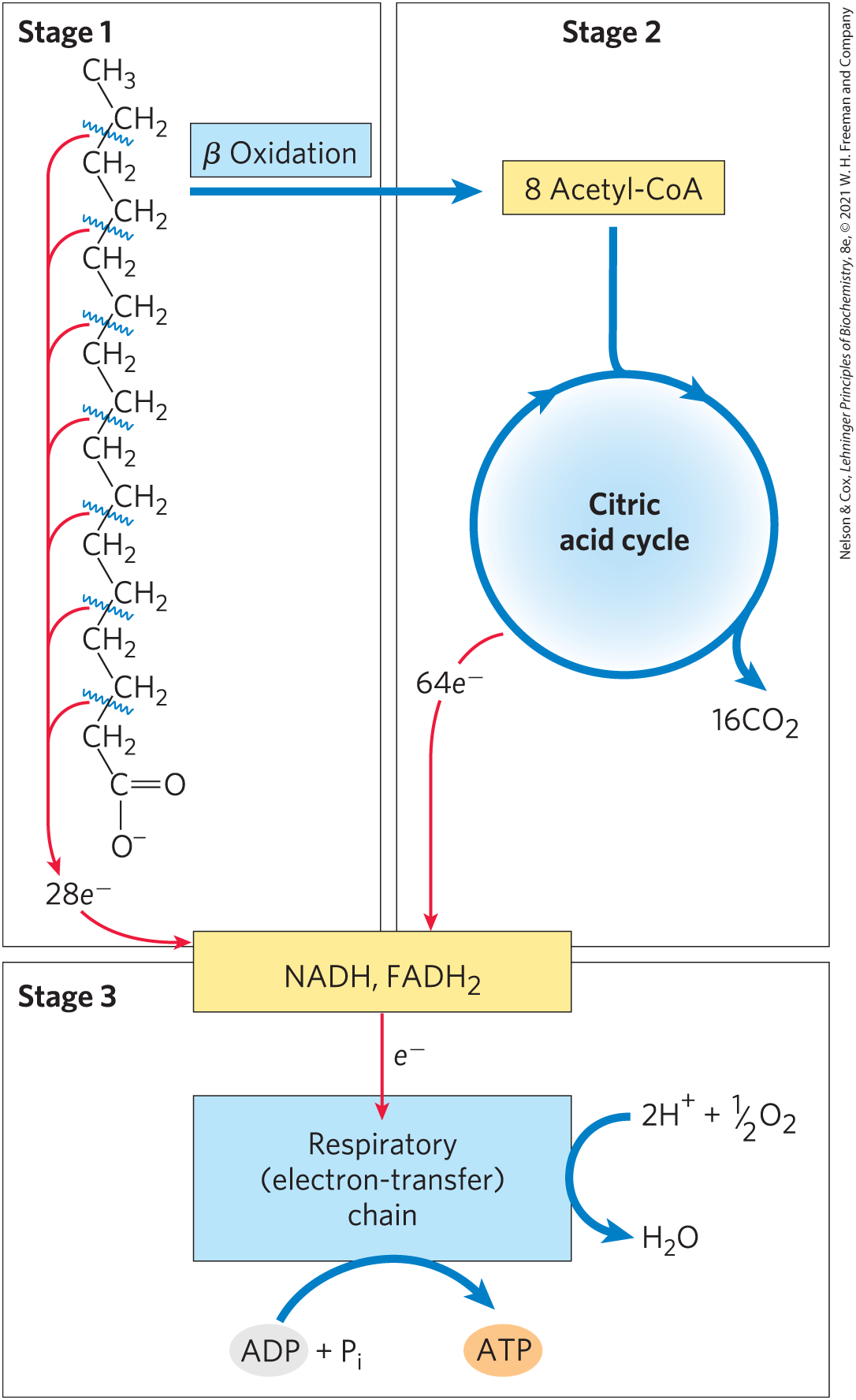
As noted earlier, mitochondrial oxidation of fatty acids takes place in three stages (Fig. 17-7). In the first stage — β oxidation — fatty acids undergo oxidative removal of successive two-carbon units in the form of acetyl-CoA, starting from the carboxyl end of the fatty acyl chain. For example, the 16-carbon palmitic acid (palmitate at pH 7) undergoes seven passes through the oxidative sequence, in each pass losing two carbons as acetyl-CoA. At the end of seven cycles, the last two carbons of palmitate (originally C-15 and C-16) remain as acetyl-CoA. The overall result is the conversion of the 16-carbon chain of palmitate to eight two-carbon acetyl groups of acetyl-CoA molecules. Formation of each acetyl-CoA requires removal of four hydrogen atoms (two pairs of electrons and four ) from the fatty acyl moiety by dehydrogenases.
FIGURE 17-7 Stages of fatty acid oxidation. Stage 1: A long-chain fatty acid is oxidized to yield acetyl residues in the form of acetyl-CoA. This process is called β oxidation. Stage 2: The acetyl groups are oxidized to via the citric acid cycle. Stage 3: Electrons derived from the oxidations of stages 1 and 2 pass to via the mitochondrial respiratory chain, providing the energy for ATP synthesis by oxidative phosphorylation.
In the second stage of fatty acid oxidation, the acetyl groups of acetyl-CoA are oxidized to in the citric acid cycle, which also takes place in the mitochondrial matrix. Acetyl-CoA derived from fatty acids thus enters a final common pathway of oxidation with the acetyl-CoA derived from glucose via glycolysis and pyruvate oxidation (see Fig. 16-1). The first two stages of fatty acid oxidation produce the reduced electron carriers NADH and , which in the third stage donate electrons to the mitochondrial respiratory chain, through which the electrons pass to oxygen with the concomitant phosphorylation of ADP to ATP (Fig. 17-7). The energy released by fatty acid oxidation is thus conserved as ATP.
We now take a closer look at the first stage of fatty acid oxidation, beginning with the simple case of a saturated fatty acyl chain with an even number of carbons, then turning to the slightly more complicated cases of unsaturated and odd-number chains. We also consider the regulation of fatty acid oxidation, the β-oxidative processes as they occur in organelles other than mitochondria, and, finally, a less-common mode of fatty acid catabolism — α oxidation.
The β Oxidation of Saturated Fatty Acids Has Four Basic Steps
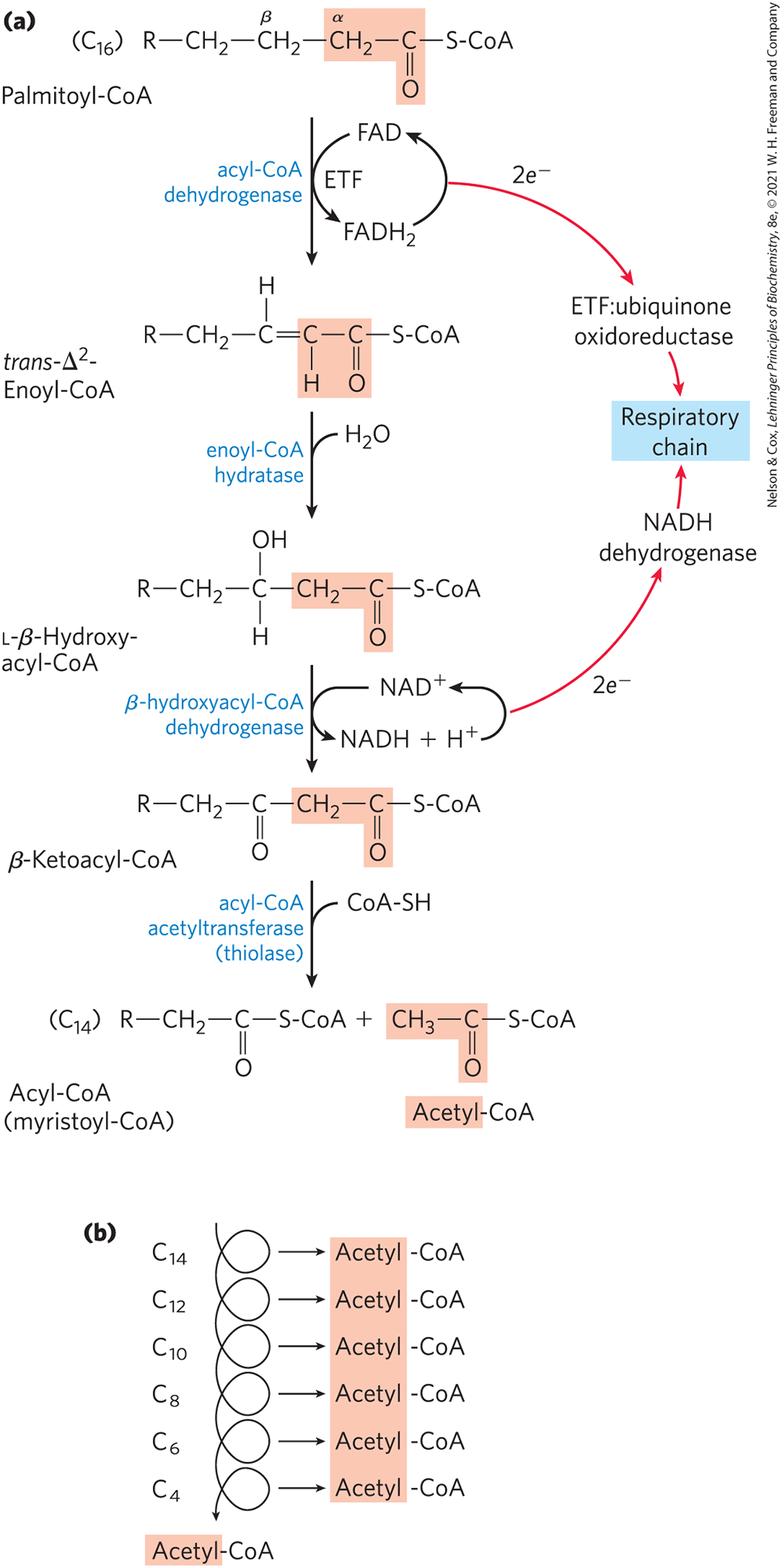
Four enzyme-catalyzed reactions make up the first stage of fatty acid oxidation (Fig. 17-8a). First, dehydrogenation of fatty acyl–CoA produces a double bond between the α and β carbon atoms (C-2 and C-3), yielding a trans--enoyl-CoA (the symbol designates the position of the double bond; you may want to review fatty acid nomenclature, p. 342). Note that the new double bond has the trans configuration, whereas the double bonds in naturally occurring unsaturated fatty acids are normally in the cis configuration. We consider the significance of this difference later.
FIGURE 17-8 The β-oxidation pathway. (a) In each pass through this four-step sequence, one acetyl residue (shaded in light red) is removed in the form of acetyl-CoA from the carboxyl end of the fatty acyl chain — in this example palmitate , which enters as palmitoyl-CoA. Electrons from the first oxidation pass through electron transfer flavoprotein (ETF), and then through a second flavoprotein (ETF:ubiquinone oxidoreductase), into the respiratory chain. Electrons from the second oxidation enter the respiratory chain through NADH dehydrogenase. (b) Six more passes through the β-oxidation pathway yield seven more molecules of acetyl-CoA, the seventh arising from the last two carbon atoms of the 16-carbon chain. Eight molecules of acetyl-CoA are formed in all. The acetyl-CoA may be oxidized in the citric acid cycle, donating more electrons to the respiratory chain.
This first step is catalyzed by three isozymes of acyl-CoA dehydrogenase, each specific for a range of fatty-acyl chain lengths: very-long-chain acyl-CoA dehydrogenase (VLCAD), acting on fatty acids of 12 to 18 carbons; medium-chain (MCAD), acting on fatty acids of 4 to 14 carbons; and short-chain (SCAD), acting on fatty acids of 4 to 8 carbons. VLCAD is in the inner mitochondrial membrane; MCAD and SCAD are in the matrix. All three isozymes are flavoproteins with tightly bound FAD (see Fig. 13-27) as a prosthetic group. The electrons removed from the fatty acyl–CoA are transferred to FAD, and the reduced form of the dehydrogenase immediately donates its electrons to an electron carrier, the electron transfer flavoprotein (ETF) (see Fig. 19-15). Electrons move from ETF to a second flavoprotein, ETF:ubiquinone oxidoreductase, and through ubiquinone into the mitochondrial respiratory chain. The oxidation catalyzed by an acyl-CoA dehydrogenase is analogous to succinate dehydrogenation in the citric acid cycle (p. 586); in both reactions the enzyme is bound to the inner membrane, a double bond is introduced into a carboxylic acid between the α and β carbons, FAD is the electron acceptor, and electrons from the reaction ultimately enter the respiratory chain and pass to , with the concomitant synthesis of about 1.5 ATP molecules per electron pair.
In the second step of the β-oxidation cycle (Fig. 17-8a), water is added to the double bond of the trans--enoyl-CoA to form the l stereoisomer of -hydroxyacyl-CoA (3-hydroxyacyl-CoA). This reaction, catalyzed by enoyl-CoA hydratase, is formally analogous to the fumarase reaction in the citric acid cycle, in which adds across an double bond (p. 587).
In the third step, l-β-hydroxyacyl-CoA is dehydrogenated to form -ketoacyl-CoA, by the action of -hydroxyacyl-CoA dehydrogenase; is the electron acceptor. This enzyme is absolutely specific for the l stereoisomer of hydroxyacyl-CoA. The NADH formed in the reaction donates its electrons to NADH dehydrogenase (Complex I), an electron carrier of the respiratory chain (see Fig. 19-15), and ATP is formed from ADP as the electrons pass to . The reaction catalyzed by β-hydroxyacyl-CoA dehydrogenase is closely analogous to the malate dehydrogenase reaction of the citric acid cycle (p. 587).
The fourth and last step of the β-oxidation cycle is catalyzed by acyl-CoA acetyltransferase, more commonly called thiolase, which promotes reaction of β-ketoacyl-CoA with a molecule of free coenzyme A to split off the carboxyl-terminal two-carbon fragment of the original fatty acid as acetyl-CoA. The other product is the coenzyme A thioester of the fatty acid, now shortened by two carbon atoms (Fig. 17-8a). This reaction is called thiolysis, by analogy with the process of hydrolysis, because the β-ketoacyl-CoA is cleaved by reaction with the thiol group of coenzyme A. The thiolase reaction is a reverse Claisen condensation (see Fig. 13-4).
The last three steps of this four-step sequence are catalyzed by either of two sets of enzymes, with the enzymes employed depending on the length of the fatty acyl chain. For fatty acyl chains of 12 or more carbons, the reactions are catalyzed by a multienzyme complex associated with the inner mitochondrial membrane, the trifunctional protein (TFP). TFP is a heterooctamer of subunits. Each α subunit contains two activities, the enoyl-CoA hydratase and the β-hydroxyacyl-CoA dehydrogenase; the β subunits contain the thiolase activity. This tight association of three enzymes allows efficient substrate channeling from one active site to the next, without diffusion of the intermediates away from the enzyme surface. When TFP has shortened the fatty acyl chain to 12 or fewer carbons, further oxidations are catalyzed by a set of four soluble enzymes in the matrix.
As noted earlier, the single bond between methylene groups in fatty acids is relatively stable. The β-oxidation sequence is an elegant mechanism for destabilizing and breaking these bonds. The first three reactions of β oxidation create a much less stable bond, in which the α carbon (C-2) is bonded to two carbonyl carbons (the β-ketoacyl-CoA intermediate). The ketone function on the β carbon (C-3) makes it a good target for nucleophilic attack by the of coenzyme A, catalyzed by thiolase. The acidity of the α hydrogen and the resonance stabilization of the carbanion generated by the departure of this hydrogen make the terminal a good leaving group, facilitating breakage of the bond.
We have already seen a reaction sequence nearly identical with these four steps of fatty acid oxidation, in the citric acid cycle reaction steps between succinate and oxaloacetate (see Fig. 16-7). A nearly identical reaction sequence occurs again in the pathways by which the branched-chain amino acids (isoleucine, leucine, and valine) are oxidized as fuels (see Fig. 18-28). Figure 17-9 shows the common features of these three sequences, almost certainly an example of the conservation of a mechanism by gene duplication and evolution of a new specificity in the enzyme products of the duplicated genes.
FIGURE 17-9 A conserved reaction sequence to introduce a carbonyl function on the carbon β to a carboxyl group. The β-oxidation pathway for fatty acyl–CoAs, the pathway from succinate to oxaloacetate in the citric acid cycle, and the pathway by which the deaminated carbon skeletons from isoleucine, leucine, and valine are oxidized as fuels — all use the same reaction sequence.
The Four β-Oxidation Steps Are Repeated to Yield Acetyl-CoA and ATP
In one pass through the β-oxidation sequence, one molecule of acetyl-CoA, two pairs of electrons, and four protons are removed from the long-chain fatty acyl–CoA, shortening it by two carbon atoms. The equation for one pass, beginning with the coenzyme A ester of our example, palmitate, is
(17-2)
Following removal of one acetyl-CoA unit from palmitoyl-CoA, the coenzyme A thioester of the shortened fatty acid (now the 14-carbon myristate) remains. The myristoyl-CoA can now go through another set of four β-oxidation reactions, exactly analogous to the first, to yield a second molecule of acetyl-CoA and lauroyl-CoA, the coenzyme A thioester of the 12-carbon laurate. Altogether, seven passes through the β-oxidation sequence are required to oxidize one molecule of palmitoyl-CoA to eight molecules of acetyl-CoA (Fig. 17-8b). The overall equation is
(17-3)
Each molecule of formed during oxidation of the fatty acid donates a pair of electrons to ETF of the respiratory chain, and about 1.5 molecules of ATP are generated during the ensuing transfer of each electron pair to . Similarly, each molecule of NADH formed delivers a pair of electrons to the mitochondrial NADH dehydrogenase, and the subsequent transfer of each pair of electrons to results in formation of about 2.5 molecules of ATP. Thus four molecules of ATP are formed for each two-carbon unit removed in one pass through the sequence.
Note that water is also produced in this process. Each pair of electrons transferred from NADH or to yields one , referred to as “metabolic water.” Reduction of by NADH also consumes one per NADH molecule: . In hibernating animals, fatty acid oxidation provides metabolic energy, heat, and water — all essential for survival of an animal that neither eats nor drinks for long periods (Box 17-1). Camels obtain water to supplement the meager supply available in their natural environment by oxidation of fats stored in their hump.
The overall equation for the oxidation of palmitoyl-CoA to eight molecules of acetyl-CoA, including the electron transfers and oxidative phosphorylations, is
(17-4)
Acetyl-CoA Can Be Further Oxidized in the Citric Acid Cycle
The acetyl-CoA produced from the oxidation of fatty acids can be oxidized to and by the citric acid cycle. The following equation represents the balance sheet for the second stage in the oxidation of palmitoyl-CoA, together with the coupled phosphorylations of the third stage:
(17-5)
Combining Equations 17-4 and 17-5, we obtain the overall equation for the complete oxidation of palmitoyl-CoA to carbon dioxide and water:
(17-6)
Table 17-1 summarizes the yields of NADH, , and ATP in the successive steps of palmitoyl-CoA oxidation. Note that because the activation of palmitate to palmitoyl-CoA breaks both phosphoanhydride bonds in ATP (Fig. 17-5), the energetic cost of activating a fatty acid is equivalent to two ATP, and the net gain per molecule of palmitate is 106 ATP. The standard free-energy change for the oxidation of palmitate to and is about 9,800 kJ/mol. Under standard conditions, the energy recovered as the phosphate bond energy of ATP is , about 33% of the theoretical maximum. However, when the free-energy changes are calculated from actual concentrations of reactants and products under intracellular conditions, the free-energy recovery is more than 60%; the energy conservation is remarkably efficient (see Worked Example 13-2, p. 480).
TABLE 17.1 Yield of ATP during Oxidation of One Molecule of Palmitoyl-CoA to and
aThese calculations assume that mitochondrial oxidative phosphorylation produces 1.5 ATP per oxidized and 2.5 ATP per NADH oxidized.
bGTP produced directly in this step yields ATP in the reaction catalyzed by nucleoside diphosphate kinase (p. 487).
Oxidation of Unsaturated Fatty Acids Requires Two Additional Reactions
The fatty acid oxidation sequence just described is typical when the incoming fatty acid is saturated (that is, has only single bonds in its carbon chain). However, most of the fatty acids in the triacylglycerols and phospholipids of animals and plants are unsaturated, having one or more double bonds. These bonds are in the cis configuration and cannot be acted upon by enoyl-CoA hydratase, the enzyme catalyzing the addition of to the trans double bond of the -enoyl-CoA generated during β oxidation. Two auxiliary enzymes — an isomerase and a reductase — must act on the common unsaturated fatty acids to transform them into substrates for the β-oxidation pathway. We illustrate these auxiliary reactions with two examples.
Oleate is an abundant 18-carbon monounsaturated fatty acid with a cis double bond between C-9 and C-10 (denoted ). In the first step of oxidation, oleate is converted to oleoyl-CoA and, like the saturated fatty acids, enters the mitochondrial matrix via the carnitine shuttle (Fig. 17-6). Oleoyl-CoA then undergoes three passes through the fatty acid oxidation cycle to yield three molecules of acetyl-CoA and the coenzyme A ester of a , 12-carbon unsaturated fatty acid, cis--dodecenoyl-CoA (Fig. 17-10). This product cannot serve as a substrate for enoyl-CoA hydratase, which acts only on trans double bonds. The auxiliary enzyme -enoyl-CoA isomerase isomerizes the cis--enoyl-CoA to the trans--enoyl-CoA, which is converted by enoyl-CoA hydratase into the corresponding l-β hydroxyacyl-CoA (trans--dodecenoyl-CoA). This intermediate is now acted upon by the remaining enzymes of β oxidation to yield acetyl-CoA and the coenzyme A ester of a 10-carbon saturated fatty acid, decanoyl-CoA. The latter undergoes four more passes through the β-oxidation pathway to yield five more molecules of acetyl-CoA. Altogether, nine acetyl-CoAs are produced from one molecule of the 18-carbon oleate.
FIGURE 17-10 Oxidation of a monounsaturated fatty acid. Oleic acid, as oleoyl-CoA , is the example used here. Oxidation requires an additional enzyme, enoyl-CoA isomerase, to reposition the double bond, converting the cis isomer to a trans isomer, an intermediate in β oxidation.
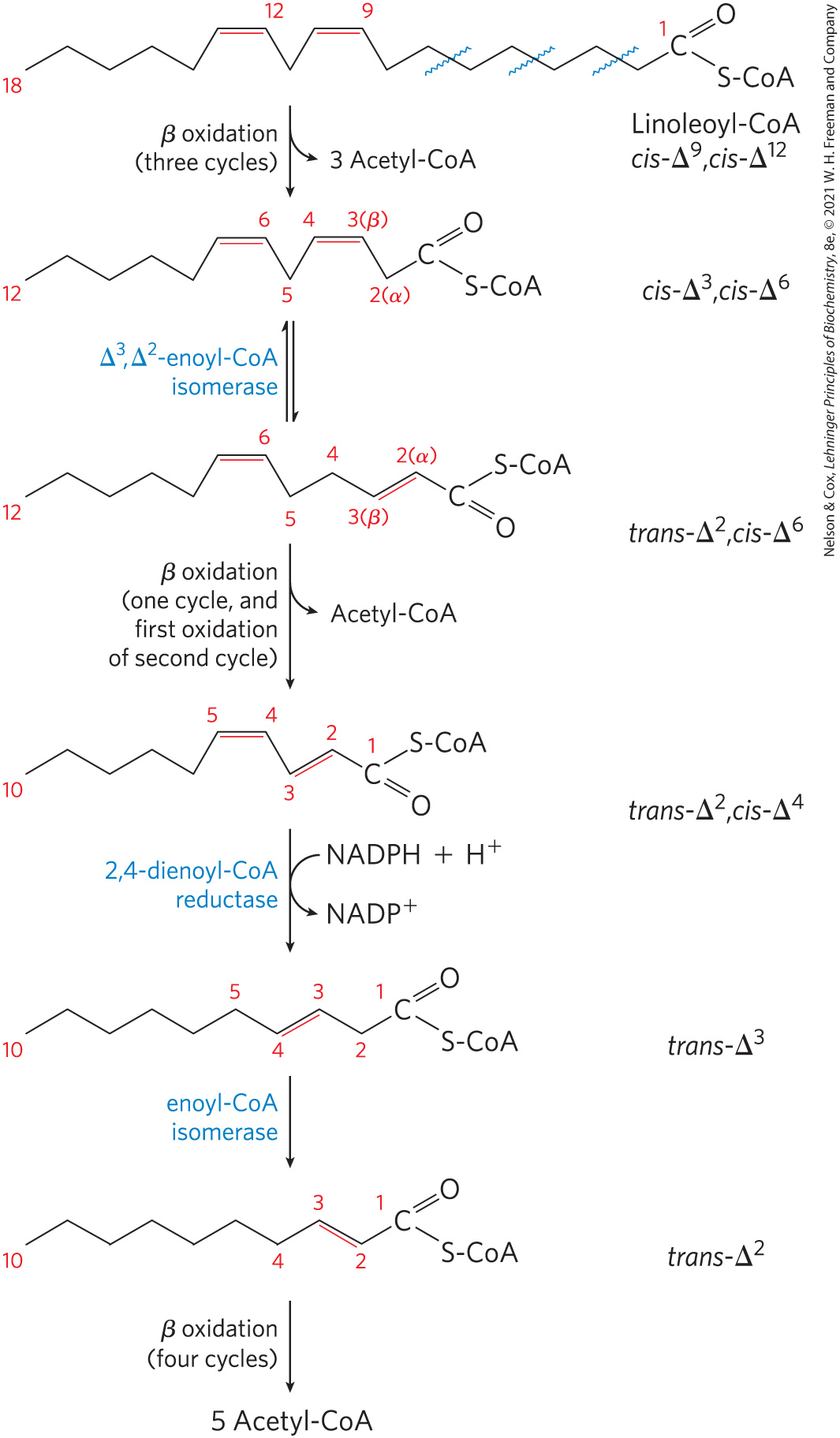
The other auxiliary enzyme (a reductase) is required for oxidation of polyunsaturated fatty acids — for example, the 18-carbon linoleate, which has a , configuration (Fig. 17-11). Linoleoyl-CoA undergoes three passes through the β-oxidation sequence to yield three molecules of acetyl-CoA and the coenzyme A ester of a 12-carbon unsaturated fatty acid with a , configuration. This intermediate cannot be used by the enzymes of the β-oxidation pathway: its double bonds are in the wrong position and have the wrong configuration (cis, not trans). However, the combined action of enoyl-CoA isomerase and 2,4-dienoyl-CoA reductase, as shown in Figure 17-11, transforms this intermediate into one that can enter the β-oxidation pathway and be degraded to six acetyl-CoAs. The overall result is conversion of linoleate to nine molecules of acetyl-CoA.
FIGURE 17-11 Oxidation of a polyunsaturated fatty acid. The example here is linoleic acid, as linoleoyl-CoA . Oxidation requires a second auxiliary enzyme in addition to enoyl-CoA isomerase: NADPH-dependent 2,4-dienoyl-CoA reductase. The combined action of these two enzymes converts a , -dienoyl-CoA intermediate to the -enoyl-CoA substrate necessary for β oxidation.
Complete Oxidation of Odd-Number Fatty Acids Requires Three Extra Reactions
Although most naturally occurring lipids contain fatty acids with an even number of carbon atoms, fatty acids with an odd number of carbons are common in the lipids of many plants and some marine organisms. Cattle and other ruminant animals form large amounts of the three-carbon propionate during fermentation of carbohydrates in the rumen. The propionate is absorbed into the blood and oxidized by the liver and other tissues.
Long-chain odd-number fatty acids are oxidized in the same pathway as the even-number acids, beginning at the carboxyl end of the chain. However, the substrate for the last pass through the β-oxidation sequence is a fatty acyl–CoA with a five-carbon fatty acid. When this is oxidized and cleaved, the products are acetyl-CoA and propionyl-CoA. The acetyl-CoA can be oxidized in the citric acid cycle, of course, but propionyl-CoA enters a different pathway, having three enzymes.
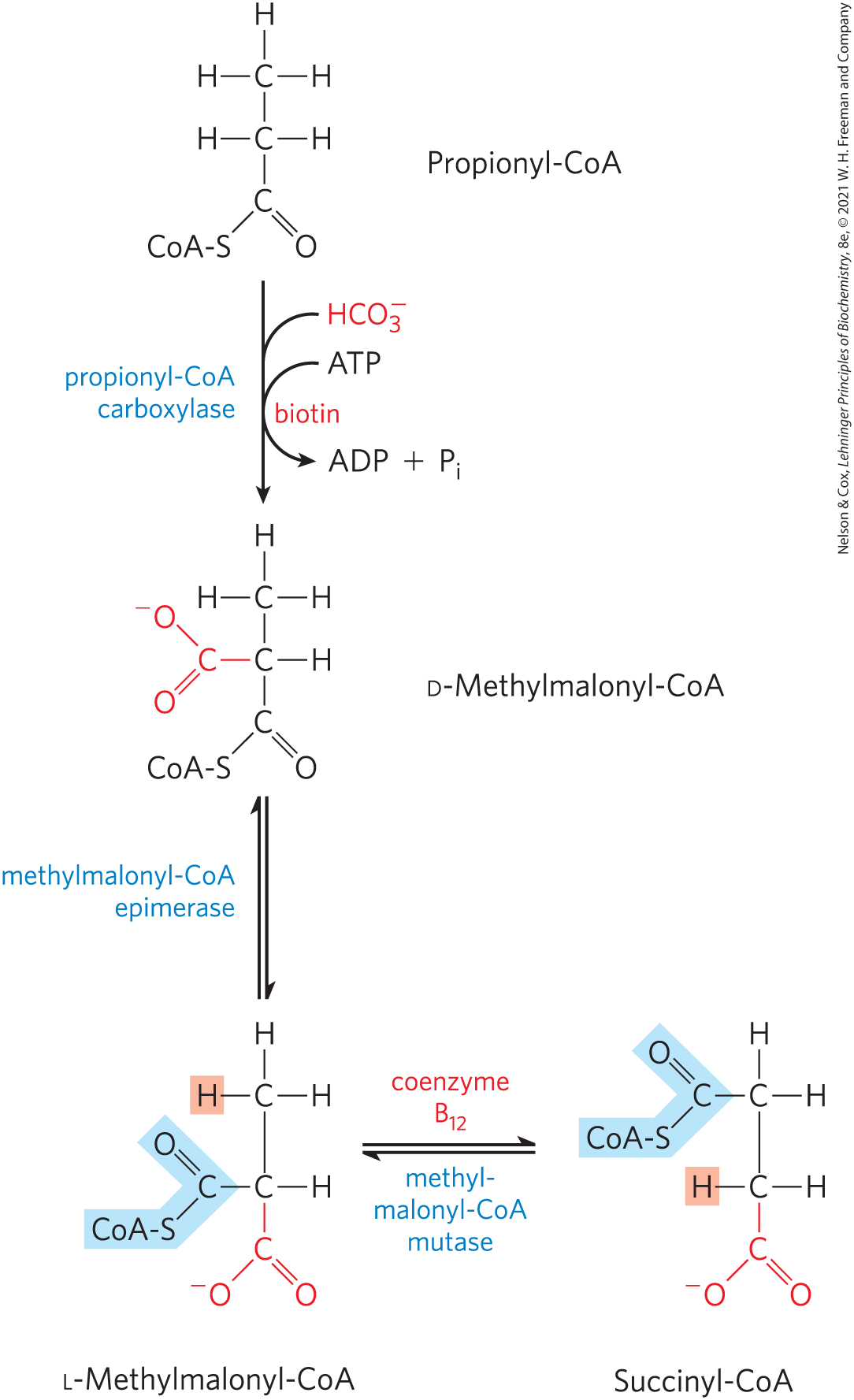
Propionyl-CoA is first carboxylated to form the d stereoisomer of methylmalonyl-CoA by propionyl-CoA carboxylase, which contains the cofactor biotin (Fig. 17-12). In this enzymatic reaction, as in the pyruvate carboxylase reaction (see Fig. 16-16), bicarbonate ion is activated by attachment to biotin before its transfer to the substrate, in this case the propionate moiety. Formation of the carboxybiotin intermediate requires energy, which is provided by ATP. The d-methylmalonyl-CoA thus formed is enzymatically epimerized to its l stereoisomer by methylmalonyl-CoA epimerase. The l-methylmalonyl-CoA then undergoes an intramolecular rearrangement to form succinyl-CoA, which can enter the citric acid cycle. This rearrangement is catalyzed by methylmalonyl-CoA mutase, which requires as its coenzyme -deoxyadenosylcobalamin, or coenzyme , which is derived from vitamin (cobalamin). Box 17-2 describes the role of coenzyme in this remarkable exchange reaction.
FIGURE 17-12 Oxidation of propionyl-CoA produced by β oxidation of odd-number fatty acids. The sequence involves the carboxylation of propionyl-CoA to d-methylmalonyl-CoA and conversion of the latter to succinyl-CoA. This conversion requires epimerization of d- to l-methylmalonyl-CoA, followed by a remarkable reaction in which substituents on adjacent carbon atoms exchange positions (see Box 17-2).
About 1 in 100,000 babies are born with a genetic deficiency in propionyl-CoA carboxylase, making them unable to metabolize the propionyl-CoA that results from catabolism of odd-number fatty acids, fatty acids with methyl branches, and the amino acids isoleucine, valine, threonine, and methionine. The absence of functional propionyl-CoA carboxylase leads to an accumulation of propionyl-CoA in mitochondria, depleting the available supply of coenzyme A for continuing β-oxidation and other metabolism. The propionyl-CoA is esterified to carnitine, transported out of the mitochondria through the carnitine shuttle, and eventually released to the blood as propionate, which severely acidifies the blood and urine, a condition called propionic acidemia. Symptoms generally manifest in the first few days of life, with vomiting, low blood glucose, seizures, and neurologic abnormalities. The condition is diagnosed by detection of propionate and its metabolites in the blood and urine. Treatments include severe restriction of dietary protein, supplying carnitine, and the use of antibiotics targeted at gut bacteria that produce odd-chain and branched-chain fatty acids. Some individuals who are defective in the incorporation of biotin into the enzyme benefit from treatment with high doses of biotin.
Fatty Acid Oxidation Is Tightly Regulated
Oxidation of fatty acids consumes a precious fuel, and it is regulated so as to occur only when the organism’s need for energy requires it. In the liver, fatty acyl–CoA formed in the cytosol has two major pathways open to it: (1) β oxidation by enzymes in mitochondria or (2) conversion into triacylglycerols and phospholipids by enzymes in the cytosol. The pathway taken depends on the rate of transfer of long-chain fatty acyl–CoA into mitochondria. The three-step carnitine shuttle by which fatty acyl groups are carried into the mitochondrial matrix as fatty acyl–carnitine (Fig. 17-6) is rate-limiting for fatty acid oxidation and is an important point of regulation. Once fatty acyl groups have entered the mitochondrion, they are committed to oxidation to acetyl-CoA.
Figure 17-13 illustrates, with numbered steps described below, the coordinated down-regulation of β oxidation when carbohydrate levels are sufficient. Ingestion of a high-carbohydrate meal raises the blood glucose level and thus triggers the release of insulin. Insulin-dependent protein phosphatase dephosphorylates the enzyme acetyl-CoA carboxylase (ACC), activating it. ACC catalyzes the formation of malonyl-CoA (the first intermediate of cytosolic fatty acid synthesis; see Figs 21-1 and 21-2), and malonyl-CoA inhibits carnitine acyltransferase 1, thereby preventing fatty acid entry into the mitochondrial matrix through the carnitine shuttle. The inhibition of carnitine acyltransferase 1 by malonyl-CoA ensures that the oxidation of fatty acids is inhibited whenever the liver is amply supplied with glucose as fuel and is actively making triacylglycerols from excess glucose. Two of the enzymes of β oxidation () are also regulated by metabolites that signal energy sufficiency. When the ratio is high, β-hydroxyacyl-CoA dehydrogenase is inhibited; in addition, high concentrations of acetyl-CoA inhibit thiolase.
FIGURE 17-13 Coordinated regulation of fatty acid synthesis and breakdown. The steps are explained in the text.
Figure 17-13 also shows that conversely, when blood glucose levels drop between meals, glucagon release activates protein kinase A (PKA), which phosphorylates and inactivates ACC. Recall from Chapter 14 that in a low-energy state, [AMP] rises relative to [ATP], activating AMP kinase (AMPK). Activated AMPK also phosphorylates and inactivates ACC. The concentration of malonyl-CoA falls, and the inhibition of fatty acid entry into mitochondria is relieved. Fatty acids enter the mitochondrial matrix, allowing β oxidation to replenish the supply of ATP. Because glucagon also triggers the mobilization of fatty acids in adipose tissue, a supply of fatty acids begins arriving in the blood.
Transcription Factors Turn on the Synthesis of Proteins for Lipid Catabolism
In addition to the various short-term regulatory mechanisms that modulate the activity of existing enzymes, transcriptional regulation can change the number of molecules of the enzymes of fatty acid oxidation on a longer time scale — minutes to hours. The PPAR family of nuclear receptors are transcription factors that affect many metabolic processes in response to a variety of fatty acid–like ligands. (They were originally recognized as peroxisome proliferator-activated receptors, then were found to function more broadly.) acts in muscle, adipose tissue, and liver to turn on a set of genes essential for fatty acid oxidation, including the fatty acid transporter, carnitine acyltransferases 1 and 2; the fatty acyl–CoA dehydrogenases for short, medium, long, and very long acyl chains; and related enzymes. This response is triggered when a cell or an organism has an increased demand for energy from fat catabolism, such as during a fast between meals or under conditions of longer-term starvation. Glucagon, released in response to low blood glucose, can act through cAMP and the transcription factor CREB to turn on certain genes for lipid catabolism.
Another situation that is accompanied by major changes in the expression of the enzymes of fatty acid oxidation is the transition from fetal to neonatal metabolism in the heart. In the fetus, the principal fuels in heart muscle are glucose and lactate, but in the neonatal heart, fatty acids are the main fuel. At the time of this transition, is activated and in turn activates the genes essential for fatty acid metabolism. As we will see in Chapter 23, two other transcription factors in the PPAR family also play crucial roles in determining the enzyme complements — and therefore the metabolic activities — of specific tissues at particular times (see Fig. 23-38).
The major sites of fatty acid oxidation, at rest and during exercise, are skeletal and heart muscle. Endurance training increases expression in muscle, leading to increased levels of fatty acid–oxidizing enzymes and increased oxidative capacity of the muscle.
Genetic Defects in Fatty Acyl–CoA Dehydrogenases Cause Serious Disease
Stored triacylglycerols are typically the chief source of energy for muscle contraction, and an inability to oxidize fatty acids from triacylglycerols has serious consequences for health. The most common genetic defect in fatty acid catabolism in U.S. and northern European populations is due to a mutation in the gene encoding the medium-chain acyl-CoA dehydrogenase (MCAD), mentioned earlier in the chapter. Among northern Europeans, the frequency of carriers (individuals with this recessive mutation on one of the two homologous chromosomes) is about 1 in 40, and about 1 individual in 10,000 has the disease — that is, has two copies of the mutant MCAD allele and is unable to oxidize fatty acids of 6 to 12 carbons. The disease is characterized by recurring episodes of a syndrome that includes fat accumulation in the liver, high blood levels of octanoic acid (8:0), low blood glucose (hypoglycemia), sleepiness, vomiting, and coma. Although individuals may have no symptoms between episodes, the episodes are very serious; mortality due to this disease is 25% to 60% in early childhood. If the genetic defect is detected shortly after birth, the infant can be started on a low-fat, high-carbohydrate diet. With early detection and careful management of the diet — including avoiding long intervals between meals, to prevent the body from turning to its fat reserves for energy — the prognosis for these individuals is good.
More than 20 human genetic defects in fatty acid transport or oxidation have been documented, most much less common than the defect in MCAD. One of the most severe disorders results from loss of the long-chain β-hydroxyacyl-CoA dehydrogenase activity of the trifunctional protein, TFP. Other disorders include defects in the α or β subunits that affect all three activities of TFP and cause serious heart disease and abnormal skeletal muscle.
Peroxisomes Also Carry Out β Oxidation
The mitochondrial matrix is the major site of fatty acid oxidation in animal cells, but other compartments also contain enzymes capable of oxidizing fatty acids to acetyl-CoA, by a pathway similar but not identical to that in mitochondria. In plant cells, the major site of β oxidation is not mitochondria but peroxisomes. In peroxisomes, organelles also found in animal cells, the intermediates for β oxidation of fatty acids are coenzyme A derivatives, and the process consists of four steps, as in mitochondrial β oxidation (Fig. 17-14): (1) dehydrogenation, (2) addition of water to the resulting double bond, (3) oxidation of the β-hydroxyacyl-CoA to a ketone, and (4) thiolytic cleavage by coenzyme A. The identical reactions also occur in glyoxysomes, organelles found only in germinating seeds.
FIGURE 17-14 Comparison of β oxidation in mitochondria and in peroxisomes and glyoxysomes. The peroxisomal/glyoxysomal system differs from the mitochondrial system in three respects: (1) the peroxisomal system prefers very-long-chain fatty acids; (2) in the first oxidative step, electrons pass directly to , generating ; and (3) the NADH formed in the second oxidative step cannot be reoxidized in the peroxisome or glyoxysome, so reducing equivalents are exported to the cytosol, eventually entering mitochondria. The acetyl-CoA produced by peroxisomes and glyoxysomes is also exported; the acetate from glyoxysomes serves as a biosynthetic precursor. Acetyl-CoA produced in mitochondria is further oxidized in the citric acid cycle.
One difference between the peroxisomal and mitochondrial pathways is in the chemistry of the first step (Fig. 17-14). In peroxisomes, the flavoprotein acyl-CoA oxidase that introduces the double bond passes electrons directly to , producing (thus the name “peroxisomes”). This strong and potentially damaging oxidant is immediately cleaved to and by catalase. Recall that in mitochondria, the electrons removed in the first oxidation step pass through the respiratory chain to to produce , and this process is accompanied by ATP synthesis. In peroxisomes, the energy released in the first oxidative step of fatty acid breakdown is not conserved as ATP, but is dissipated as heat.
A second important difference between mitochondrial and peroxisomal β oxidation in mammals is in the specificity for fatty acyl–CoAs; the peroxisomal system is much more active on very-long-chain fatty acids such as hexacosanoic acid (26:0) and on branched-chain fatty acids such as phytanic acid and pristanic acid (see Fig. 17-15). These less-common fatty acids are obtained from dietary intake of dairy products, the fat of ruminant animals, meat, and fish. Their catabolism in the peroxisome involves several auxiliary enzymes unique to this organelle. The inability to oxidize these compounds is responsible for several serious human genetic diseases. Individuals with Zellweger syndrome are unable to make peroxisomes and therefore lack all the metabolism unique to that organelle. In X-linked adrenoleukodystrophy (XALD), peroxisomes fail to oxidize very-long-chain fatty acids, apparently due to lack of a functional transporter for these fatty acids in the peroxisomal membrane. Both defects lead to accumulation in the blood of very-long-chain fatty acids, especially 26:0. XALD affects young boys before the age of 10 years, causing loss of vision, behavioral disturbances, and death within a few years.
In mammals, high concentrations of fats in the diet result in increased synthesis of the enzymes of peroxisomal β oxidation in the liver. Liver peroxisomes do not contain the enzymes of the citric acid cycle and cannot catalyze the oxidation of acetyl-CoA to . Instead, long-chain or branched fatty acids are catabolized in peroxisomes to shorter-chain products, such as hexanoyl-CoA, which are exported to mitochondria and completely oxidized there. As we will see in Chapter 20, the germinating seeds of plants can synthesize carbohydrates and many other metabolites from acetyl-CoA produced in peroxisomes, using a pathway (the glyoxylate cycle) not present in vertebrates.
Phytanic Acid Undergoes α Oxidation in Peroxisomes
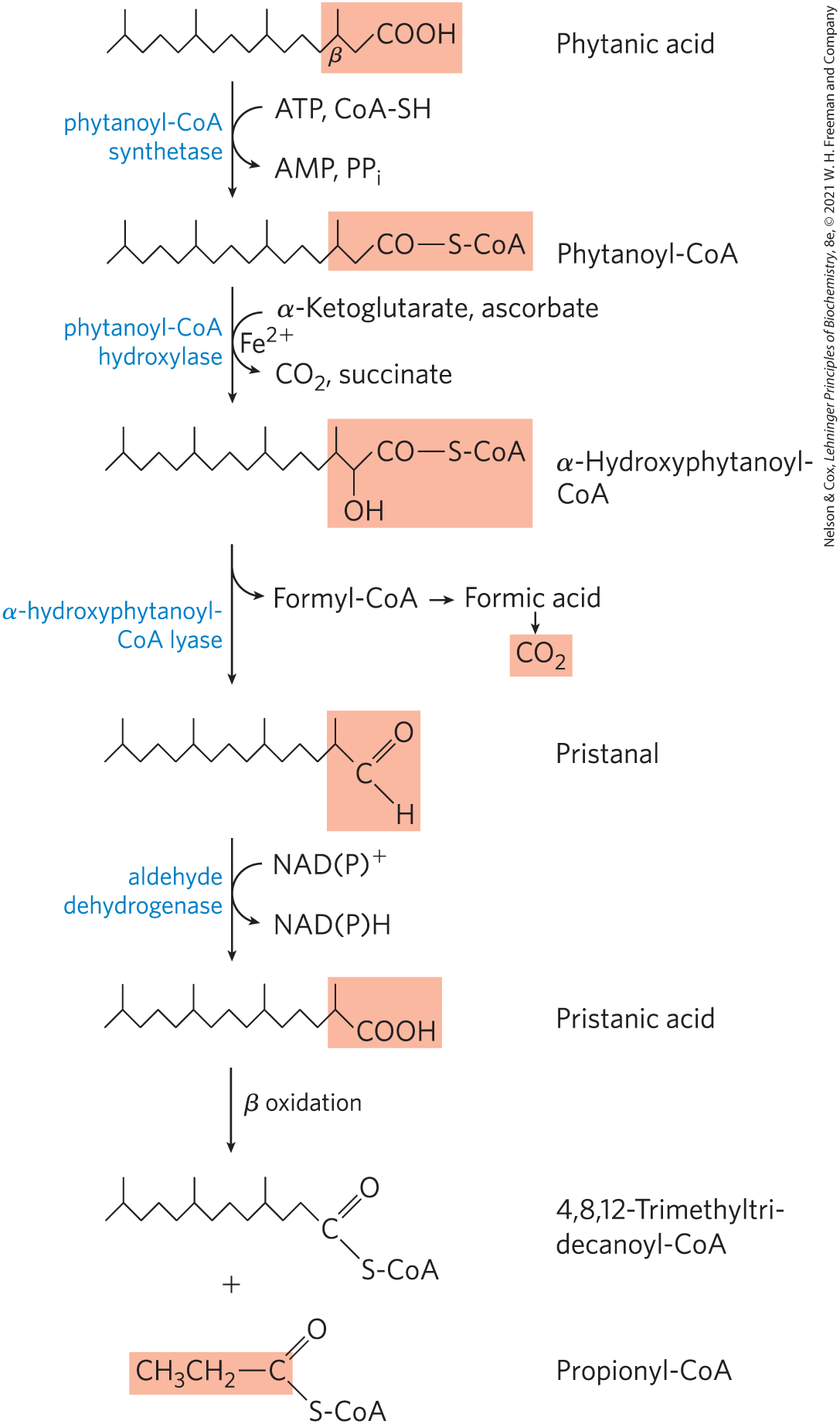
Phytanic acid, a long-chain fatty acid with methyl branches, is derived from the phytol side chain of chlorophyll (see Fig. 20-5). The presence of a methyl group on the β carbon of this fatty acid prevents the formation of a β-keto intermediate, making its β oxidation impossible. Humans obtain phytanic acid in the diet, primarily from dairy products and from the fats of ruminant animals; microorganisms in the rumen of these animals produce phytanic acid as they digest plant chlorophyll. The typical western diet includes 50 to 100 mg of phytanic acid per day.
Phytanic acid is metabolized in peroxisomes by α oxidation, in which a single carbon is removed from the carboxyl end of the fatty acid (Fig. 17-15). Phytanoyl-CoA is first hydroxylated on its α carbon in a reaction that involves molecular oxygen. The product is decarboxylated to form an aldehyde one carbon shorter, and then oxidized to the corresponding carboxylic acid, which now has no substituent on the β carbon. Further β oxidation produces acetyl-CoA and then propionyl-CoA in successive oxidation cycles. Refsum disease, resulting from a genetic defect in phytanoyl-CoA hydroxylase, leads to the accumulation of very high blood levels of phytanic acid, causing (by unknown mechanisms) severe neurological deficits, including blindness and deafness.
FIGURE 17-15 The α oxidation of a branched-chain fatty acid (phytanic acid) in peroxisomes. Phytanic acid has a methyl-substituted β carbon and therefore cannot undergo β oxidation. The combined action of the enzymes shown here removes the carboxyl carbon of phytanic acid to produce pristanic acid, in which the β carbon is unsubstituted, allowing β oxidation. Notice that β oxidation of pristanic acid releases propionyl-CoA, not acetyl-CoA. This is further catabolized as in Figure 17-12. (The details of the reaction that produces pristanal remain controversial.)
SUMMARY 17.2 Oxidation of Fatty Acids
In the first stage of β oxidation, four sequential reactions remove each acetyl-CoA unit, in turn, from the carboxyl end of a saturated fatty acyl–CoA: (1) dehydrogenation of the α and β carbons by acyl–CoA dehydrogenases; (2) hydration of the resulting trans- double bond by enoyl-CoA hydratase; (3) dehydrogenation of the resulting l-β-hydroxyacyl-CoA; and (4) cleavage of the resulting β-ketoacyl–CoA by thiolase, to form acetyl-CoA and a fatty acyl–CoA shortened by two carbons.
The shortened fatty acyl–CoA then reenters the β oxidation sequence for removal of another, and then another, acetyl-CoA.
In the second stage of fatty acid oxidation, the acetyl-CoA is oxidized to in the citric acid cycle. Much of the free energy from fatty acid oxidation is recovered as ATP by oxidative phosphorylation, the final stage of the oxidative pathway.
Oxidation of unsaturated fatty acids requires two additional enzymes: enoyl-CoA isomerase and 2,4-dienoyl-CoA reductase.
Odd-number fatty acids are oxidized by the β-oxidation pathway to yield acetyl-CoA and a molecule of propionyl-CoA. The propionyl-CoA is carboxylated to methylmalonyl-CoA, which is isomerized to succinyl-CoA in a reaction catalyzed by methylmalonyl-CoA mutase, an enzyme requiring coenzyme .
To prevent futile cycling, entry of fatty acids into mitochondria is blocked by the first intermediate in fatty acid synthesis, malonyl-CoA.
The transcription factor stimulates the synthesis of several enzymes required in β oxidation when glucose is not available as an energy source.
Individuals who lack medium-chain acyl-CoA dehydrogenase experience fatty liver, high blood levels of octanoic acid, coma, and sometimes death.
Peroxisomes, of plants and animals, and glyoxysomes, of plants, carry out β oxidation in four steps similar to those of the mitochondrial pathway. The first oxidation step, however, transfers electrons directly to , generating . Peroxisomes of animal tissues specialize in the oxidation of very-long-chain fatty acids and branched fatty acids.
The reactions of α oxidation convert branched fatty acids such as phytanic acid to products that can undergo β oxidation, yielding acetyl-CoA and propionyl-CoA.
 The oxidation catalyzed by an acyl-CoA dehydrogenase is analogous to succinate dehydrogenation in the citric acid cycle (
The oxidation catalyzed by an acyl-CoA dehydrogenase is analogous to succinate dehydrogenation in the citric acid cycle (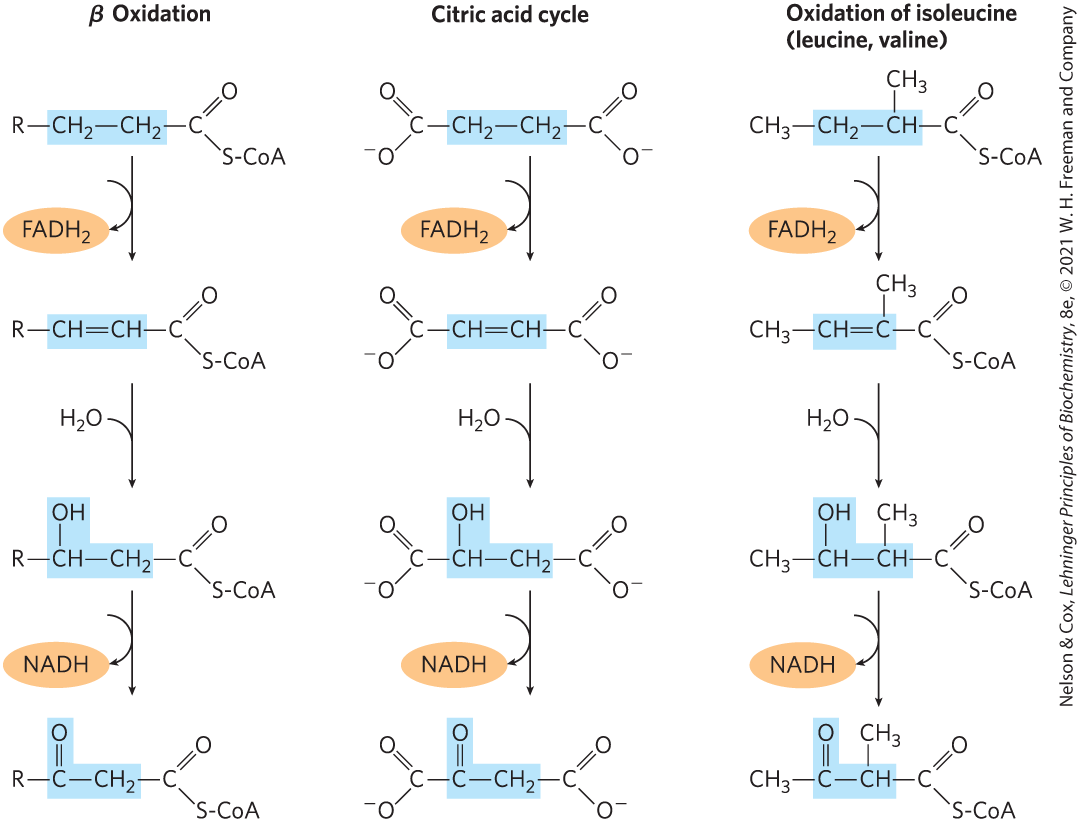
 Each molecule of formed during oxidation of the fatty acid donates a pair of electrons to ETF of the respiratory chain, and about 1.5 molecules of ATP are generated during the ensuing transfer of each electron pair to . Similarly, each molecule of NADH formed delivers a pair of electrons to the mitochondrial NADH dehydrogenase, and the subsequent transfer of each pair of electrons to results in formation of about 2.5 molecules of ATP. Thus four molecules of ATP are formed for each two-carbon unit removed in one pass through the sequence.
Each molecule of formed during oxidation of the fatty acid donates a pair of electrons to ETF of the respiratory chain, and about 1.5 molecules of ATP are generated during the ensuing transfer of each electron pair to . Similarly, each molecule of NADH formed delivers a pair of electrons to the mitochondrial NADH dehydrogenase, and the subsequent transfer of each pair of electrons to results in formation of about 2.5 molecules of ATP. Thus four molecules of ATP are formed for each two-carbon unit removed in one pass through the sequence.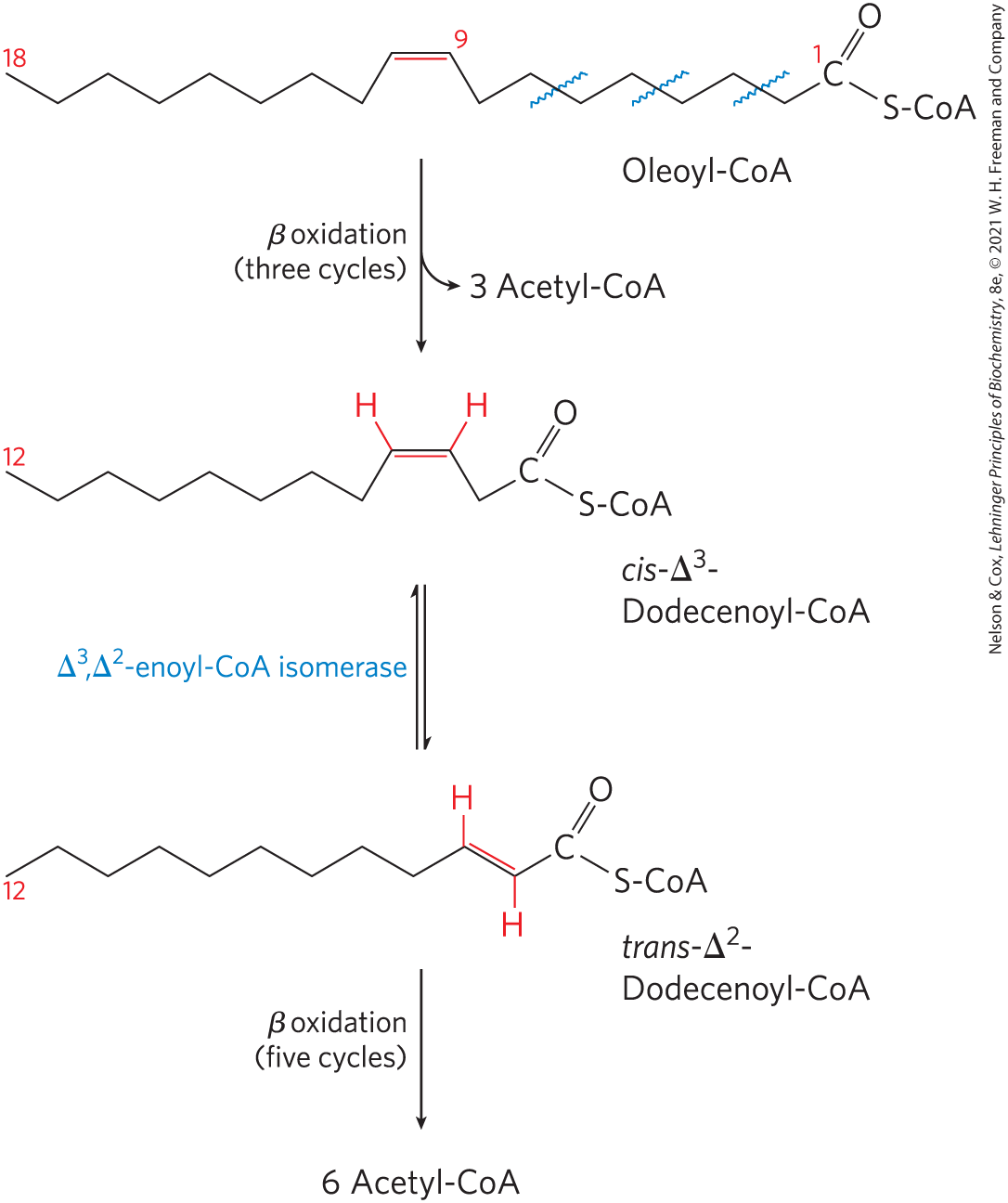
 About 1 in 100,000 babies are born with a genetic deficiency in propionyl-CoA carboxylase, making them unable to metabolize the propionyl-CoA that results from catabolism of odd-number fatty acids, fatty acids with methyl branches, and the amino acids isoleucine, valine, threonine, and methionine.
About 1 in 100,000 babies are born with a genetic deficiency in propionyl-CoA carboxylase, making them unable to metabolize the propionyl-CoA that results from catabolism of odd-number fatty acids, fatty acids with methyl branches, and the amino acids isoleucine, valine, threonine, and methionine.  The absence of functional propionyl-CoA carboxylase leads to an accumulation of propionyl-CoA in mitochondria, depleting the available supply of coenzyme A for continuing β-oxidation and other metabolism. The propionyl-CoA is esterified to carnitine, transported out of the mitochondria through the carnitine shuttle, and eventually released to the blood as propionate, which severely acidifies the blood and urine, a condition called propionic acidemia. Symptoms generally manifest in the first few days of life, with vomiting, low blood glucose, seizures, and neurologic abnormalities. The condition is diagnosed by detection of propionate and its metabolites in the blood and urine. Treatments include severe restriction of dietary protein, supplying carnitine, and the use of antibiotics targeted at gut bacteria that produce odd-chain and branched-chain fatty acids. Some individuals who are defective in the incorporation of biotin into the enzyme benefit from treatment with high doses of biotin.
The absence of functional propionyl-CoA carboxylase leads to an accumulation of propionyl-CoA in mitochondria, depleting the available supply of coenzyme A for continuing β-oxidation and other metabolism. The propionyl-CoA is esterified to carnitine, transported out of the mitochondria through the carnitine shuttle, and eventually released to the blood as propionate, which severely acidifies the blood and urine, a condition called propionic acidemia. Symptoms generally manifest in the first few days of life, with vomiting, low blood glucose, seizures, and neurologic abnormalities. The condition is diagnosed by detection of propionate and its metabolites in the blood and urine. Treatments include severe restriction of dietary protein, supplying carnitine, and the use of antibiotics targeted at gut bacteria that produce odd-chain and branched-chain fatty acids. Some individuals who are defective in the incorporation of biotin into the enzyme benefit from treatment with high doses of biotin. 
 Oxidation of fatty acids consumes a precious fuel, and it is regulated so as to occur only when the organism’s need for energy requires it. In the liver, fatty acyl–CoA formed in the cytosol has two major pathways open to it: (1) β oxidation by enzymes in mitochondria or (2) conversion into triacylglycerols and phospholipids by enzymes in the cytosol. The pathway taken depends on the rate of transfer of long-chain fatty acyl–CoA into mitochondria. The three-step carnitine shuttle by which fatty acyl groups are carried into the mitochondrial matrix as fatty acyl–carnitine (
Oxidation of fatty acids consumes a precious fuel, and it is regulated so as to occur only when the organism’s need for energy requires it. In the liver, fatty acyl–CoA formed in the cytosol has two major pathways open to it: (1) β oxidation by enzymes in mitochondria or (2) conversion into triacylglycerols and phospholipids by enzymes in the cytosol. The pathway taken depends on the rate of transfer of long-chain fatty acyl–CoA into mitochondria. The three-step carnitine shuttle by which fatty acyl groups are carried into the mitochondrial matrix as fatty acyl–carnitine ( triggers the release of insulin.
triggers the release of insulin.  Insulin-dependent protein phosphatase dephosphorylates the enzyme acetyl-CoA carboxylase (ACC), activating it.
Insulin-dependent protein phosphatase dephosphorylates the enzyme acetyl-CoA carboxylase (ACC), activating it.  ACC catalyzes the formation of malonyl-CoA (the first intermediate of cytosolic fatty acid synthesis; see
ACC catalyzes the formation of malonyl-CoA (the first intermediate of cytosolic fatty acid synthesis; see  malonyl-CoA inhibits carnitine acyltransferase 1, thereby preventing fatty acid entry into the mitochondrial matrix through the carnitine shuttle. The inhibition of carnitine acyltransferase 1 by malonyl-CoA ensures that the oxidation of fatty acids is inhibited whenever the liver is amply supplied with glucose as fuel and is actively making triacylglycerols from excess glucose. Two of the enzymes of β oxidation (
malonyl-CoA inhibits carnitine acyltransferase 1, thereby preventing fatty acid entry into the mitochondrial matrix through the carnitine shuttle. The inhibition of carnitine acyltransferase 1 by malonyl-CoA ensures that the oxidation of fatty acids is inhibited whenever the liver is amply supplied with glucose as fuel and is actively making triacylglycerols from excess glucose. Two of the enzymes of β oxidation ( ) are also regulated by metabolites that signal energy sufficiency. When the ratio is high, β-hydroxyacyl-CoA dehydrogenase is inhibited; in addition, high concentrations of acetyl-CoA inhibit thiolase.
) are also regulated by metabolites that signal energy sufficiency. When the ratio is high, β-hydroxyacyl-CoA dehydrogenase is inhibited; in addition, high concentrations of acetyl-CoA inhibit thiolase.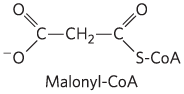
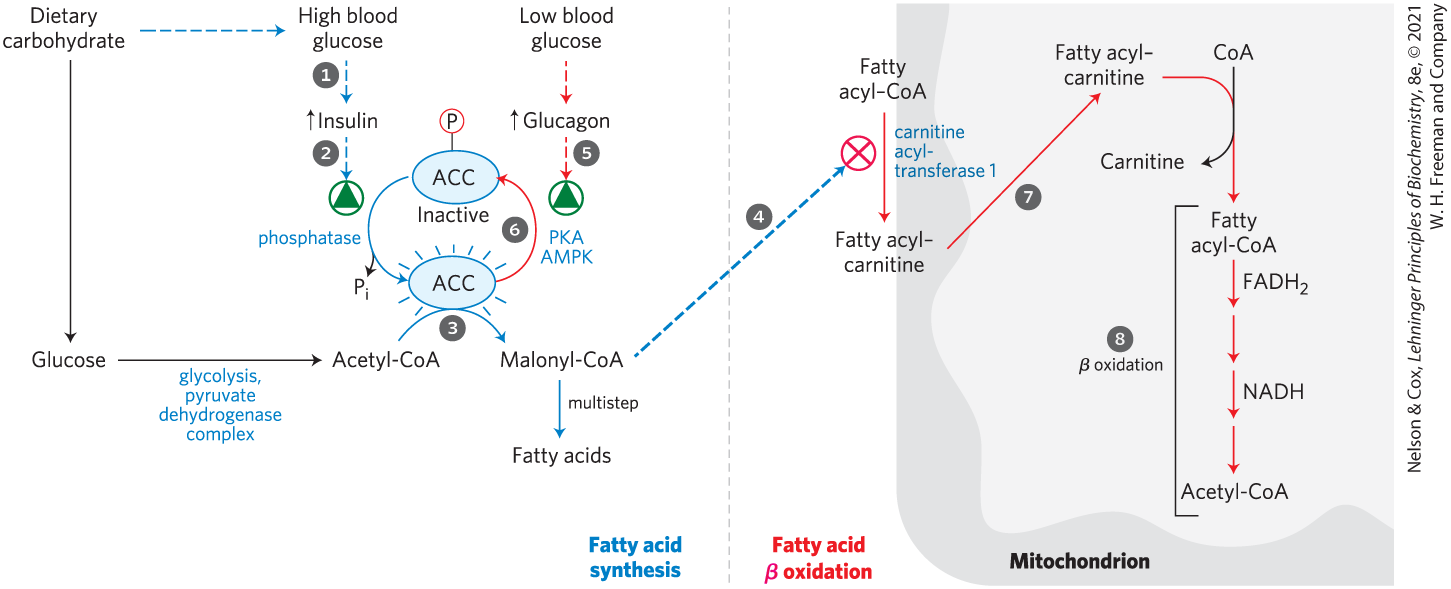
 glucagon release activates protein kinase A (PKA), which
glucagon release activates protein kinase A (PKA), which  phosphorylates and inactivates ACC. Recall from
phosphorylates and inactivates ACC. Recall from  enter the mitochondrial matrix,
enter the mitochondrial matrix, 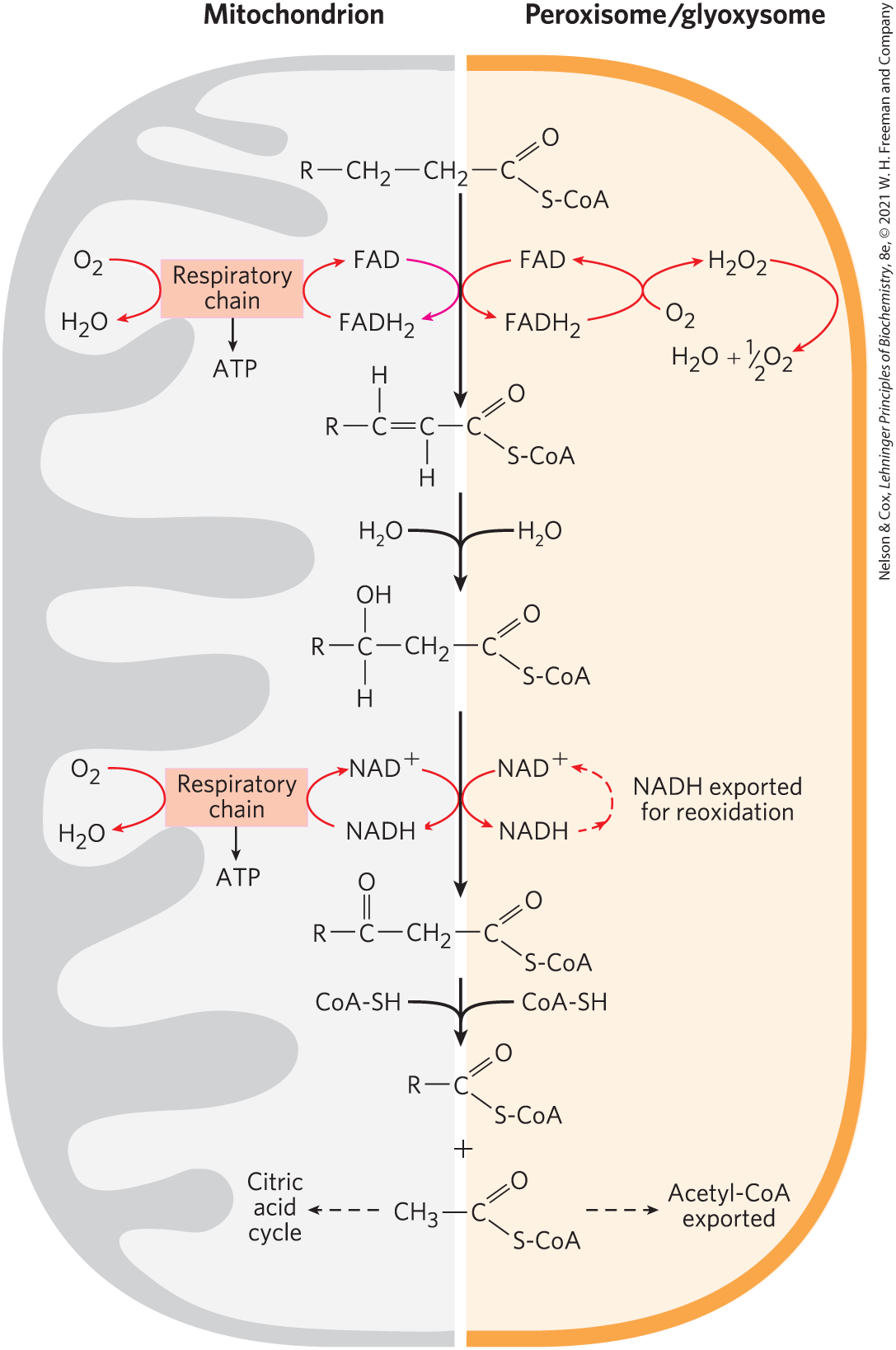
 In the first stage of β oxidation, four sequential reactions remove each acetyl-CoA unit, in turn, from the carboxyl end of a saturated fatty acyl–CoA: (1) dehydrogenation of the α and β carbons by acyl–CoA dehydrogenases; (2) hydration of the resulting trans- double bond by enoyl-CoA hydratase; (3) dehydrogenation of the resulting
In the first stage of β oxidation, four sequential reactions remove each acetyl-CoA unit, in turn, from the carboxyl end of a saturated fatty acyl–CoA: (1) dehydrogenation of the α and β carbons by acyl–CoA dehydrogenases; (2) hydration of the resulting trans- double bond by enoyl-CoA hydratase; (3) dehydrogenation of the resulting