23.4 Obesity and the Regulation of Body Mass
In the U.S. population, more than 40% of adults are obese, including more than 10% who are severely obese, as defined in terms of body mass index (BMI), calculated as . A BMI below 25 is considered normal; an individual with a BMI of 25 to 30 is overweight; a BMI greater than 30 indicates obesity; a BMI greater than 40 indicates severe obesity. Obesity is life-threatening. It significantly increases the likelihood of developing type 2 diabetes, as well as heart attack, stroke, and cancers of the colon, breast, prostate, and endometrium. Consequently, there is great interest in understanding how body mass and the storage of fats in adipose tissue are regulated.
 In the U.S. population, more than 40% of adults are obese, including more than 10% who are severely obese, as defined in terms of
In the U.S. population, more than 40% of adults are obese, including more than 10% who are severely obese, as defined in terms of 
To a first approximation, obesity is the result of taking in more calories in the diet than are expended by the body’s fuel-consuming activities. The body can deal with an excess of dietary calories in three ways: (1) convert excess fuel to fat and store it in adipose tissue, (2) burn excess fuel by extra exercise, and (3) “waste” fuel by diverting it to heat production (thermogenesis) by uncoupled mitochondria. In mammals, a complex set of hormonal and neuronal signals acts to keep fuel intake and energy expenditure in balance so as to hold the amount of adipose tissue at a suitable level. Dealing effectively with obesity requires understanding how the various checks and balances work under normal conditions and how these homeostatic mechanisms can fail.
 In mammals,
In mammals,Adipose Tissue Has Important Endocrine Functions
One early hypothesis to explain body-mass homeostasis, the “adiposity negative-feedback” model, postulated a mechanism that inhibits eating behavior and increases energy expenditure whenever body weight exceeds a certain value, called the set point; the inhibition is relieved when body weight drops below the set point (Fig. 23-29). This model predicts that a feedback signal originating in adipose tissue influences the brain centers that control eating behavior and metabolic and motor activity. The first such signal to be discovered was leptin, in 1994. Subsequent research revealed that adipose tissue is an important endocrine organ that produces peptide hormones, known as adipokines. Adipokines may act locally (autocrine and paracrine action) or systemically (endocrine action), carrying information about the adequacy of the energy reserves (TAGs) stored in adipose tissue to other tissues and to the brain. Normally, adipokines produce changes in fuel metabolism and feeding behavior that reestablish adequate fuel reserves and maintain body mass. When adipokines are over- or underproduced, the resulting dysregulation may result in life-threatening disease.
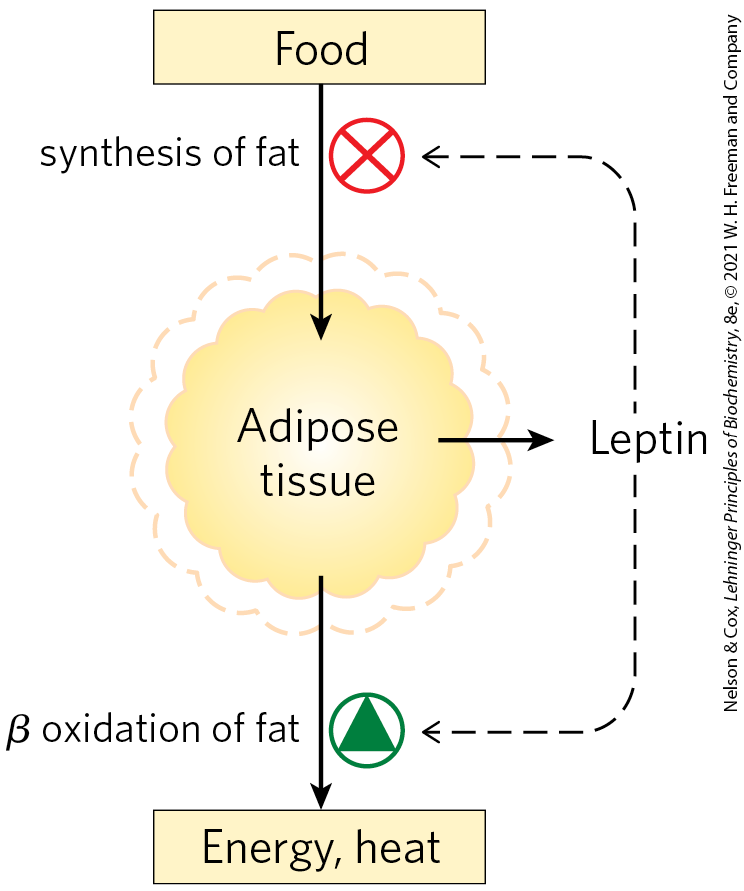
FIGURE 23-29 Set-point model for maintaining constant mass. When the mass of adipose tissue increases (dashed outline), released leptin inhibits feeding and fat synthesis and stimulates oxidation of fatty acids. When the mass of adipose tissue decreases (solid outline), lowered leptin production favors greater food intake and less fatty acid oxidation.
Leptin (Greek leptos, “thin”) is an adipokine (167 amino acid residues) that, on reaching the brain, acts on receptors in the hypothalamus to curtail appetite. Leptin was first identified as the product of a gene designated OB (obese) in laboratory mice. Mice with two defective copies of this gene (ob/ob genotype; lowercase letters signify a mutant form of the gene) show the behavior and physiology of animals in a constant state of starvation: their plasma cortisol levels are elevated; they exhibit unrestrained appetite, are unable to stay warm, grow abnormally large, and do not reproduce. As a consequence of unrestrained appetite, they become severely obese, weighing as much as three times more than normal mice (Fig. 23-30). They also have metabolic disturbances similar to those seen in diabetes, and they are insulin-resistant. When leptin is injected into ob/ob mice, they eat less, lose weight, and increase their locomotor activity and thermogenesis.

FIGURE 23-30 Obesity caused by defective leptin production. Both of these mice, which are the same age, have defects in the OB gene. The mouse on the right was injected daily with purified leptin and weighs 35 g. The mouse on the left got no leptin and consequently ate more food and was less active; it weighs 67 g.
A second mouse gene, designated DB (diabetic), also has a role in appetite regulation. Mice with two defective copies (db/db) are obese and diabetic. The DB gene encodes the leptin receptor. When the receptor is defective, the signaling function of leptin is lost.
The leptin receptor is expressed primarily in regions of the brain known to regulate feeding behavior — neurons of the arcuate nucleus of the hypothalamus (Fig. 23-31a). Leptin carries the message that fat reserves are sufficient, and it promotes reduction of fuel intake and increase in expenditure of energy. Leptin-receptor interaction in the hypothalamus alters the release of neuronal signals to the region of the brain that affects appetite. Leptin also stimulates the sympathetic nervous system, increasing blood pressure, heart rate, and thermogenesis by uncoupling the mitochondria of brown adipocytes (Fig. 23-31b). Recall that the uncoupling protein UCP1 forms a channel in the inner mitochondrial membrane that allows protons to reenter the mitochondrial matrix without passing through the ATP synthase complex. This permits constant oxidation of fuel (fatty acids in a brown or beige adipocyte) without ATP synthesis, dissipating energy as heat and consuming dietary calories or stored fats in potentially large amounts.
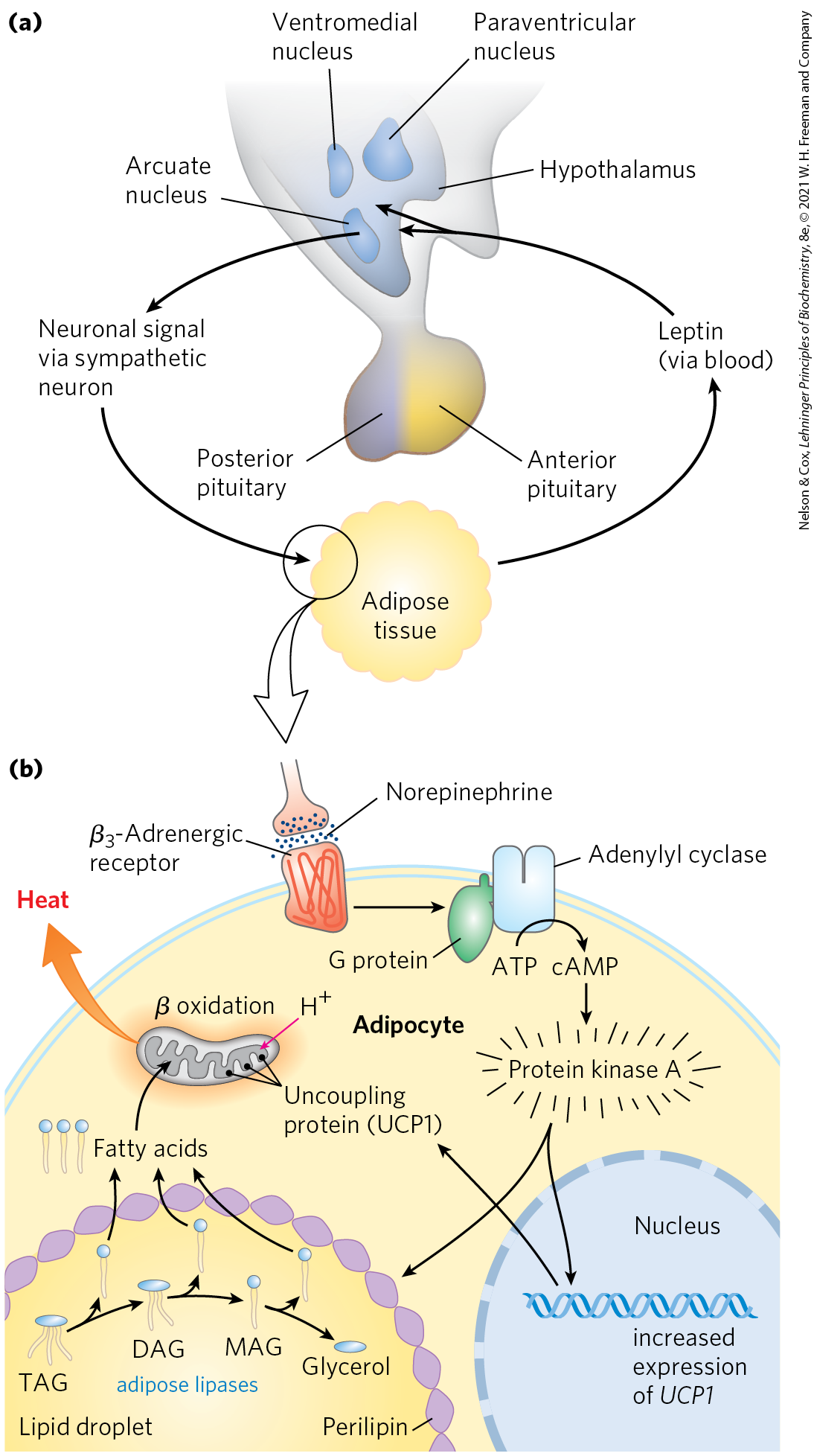
FIGURE 23-31 Hypothalamic regulation of food intake and energy expenditure. (a) Role of the hypothalamus in its interaction with adipose tissue. The hypothalamus receives input (leptin) from adipose tissue and responds with neuronal signals to adipocytes. (b) This signal (norepinephrine) activates protein kinase A, which triggers mobilization of fatty acids from TAG and their uncoupled oxidation in mitochondria, generating heat but not ATP. DAG, diacylglycerol; MAG, monoacylglycerol.
Leptin Stimulates Production of Anorexigenic Peptide Hormones
Two types of neurons in the arcuate nucleus control fuel intake and metabolism (Fig. 23-32). The orexigenic (appetite-stimulating) neurons stimulate eating by producing and releasing neuropeptide Y (NPY), which causes the next neuron in the circuit to send the signal to the brain: Eat! The blood level of NPY rises during starvation and is elevated in mice with either two defective copies of the leptin gene (ob/ob) or two defective copies of the leptin receptor gene (db/db). The high NPY concentration presumably contributes to the obesity of these mice, which eat voraciously.
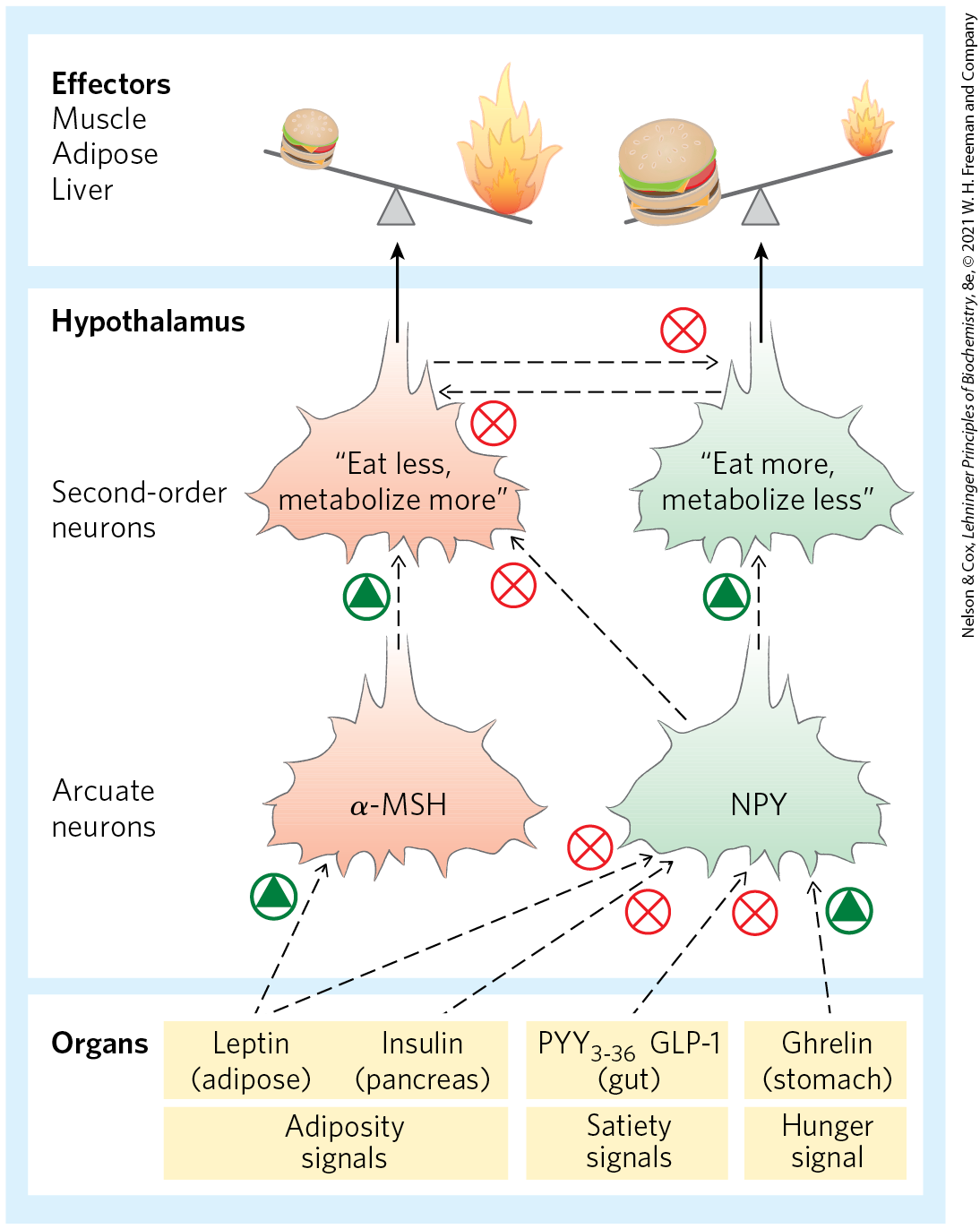
FIGURE 23-32 Hormones that control eating. In the arcuate nucleus of the hypothalamus, two sets of neurosecretory cells receive hormonal input and relay neuronal signals to the cells of muscle, adipose tissue, and liver. Leptin and insulin are released from adipose tissue and the pancreas, respectively, in proportion to the mass of body fat. The two hormones act on anorexigenic neurosecretory cells to trigger release of α-MSH (melanocyte-stimulating hormone); α-MSH carries the signal to second-order neurons in the hypothalamus, which puts out the signals to eat less and metabolize more fuel. Leptin and insulin also act on orexigenic neurosecretory cells to inhibit the release of NPY, reducing the “eat more” signal sent to the tissues. As described later in the text, the gastric hormone ghrelin stimulates appetite by activating the NPY-expressing cells; , released from the colon, inhibits these neurons and decreases appetite. Each of the two types of neurosecretory cells inhibits hormone production by the other, so any stimulus that activates orexigenic cells inactivates anorexigenic cells, and vice versa. This strengthens the effect of stimulatory inputs.
The anorexigenic (appetite-suppressing) neurons in the arcuate nucleus produce α-melanocyte–stimulating hormone (α-MSH; also known as melanocortin), formed from its polypeptide precursor pro-opiomelanocortin (POMC; Fig. 23-5). Release of α-MSH causes the next neuron in the circuit to send the signal to the brain: Stop eating!
The amount of leptin released by adipose tissue depends on both the number and the size of adipocytes. When weight loss decreases the mass of lipid tissue, leptin levels in the blood decrease, the production of NPY increases, and the processes in adipose tissue shown in Figure 23-31 are reversed. Uncoupling is diminished, slowing thermogenesis and saving fuel, and fat mobilization slows in response to reduced signaling by cAMP. Consumption of more food, combined with more efficient utilization of fuel, results in replenishment of the fat reserve in adipose tissue, bringing the system back into balance.
Leptin Triggers a Signaling Cascade That Regulates Gene Expression
The leptin signal is transduced by a mechanism also used by receptors for some growth factors. The leptin receptor has a single transmembrane segment. When leptin binds to the extracellular domains of two monomers, they dimerize and undergo phosphorylation on several Tyr residues. This initiates a chain of events that ends in the nucleus with the increased synthesis of target genes including the gene for POMC, from which α-MSH is produced.
The increased catabolism and thermogenesis triggered by leptin are due in part to increased synthesis of the mitochondria in brown and beige adipocytes. Leptin stimulates UCP1 synthesis by altering synaptic transmissions from neurons of the arcuate nucleus to adipose and other tissues via the sympathetic nervous system. The consequent increased release of norepinephrine in these tissues acts through -adrenergic receptors to stimulate transcription of the UCP1 gene. The resulting uncoupling of electron transfer from oxidative phosphorylation consumes fat and is thermogenic (Fig. 23-31).
Might human obesity be the result of insufficient leptin production and therefore be treatable by the injection of leptin? Blood levels of leptin are, in fact, usually much higher in obese animals (including humans) than in animals of normal body mass (except, of course, in ob/ob mutants, which cannot make leptin). Some downstream element in the leptin response system must be defective in obese individuals, and the elevation in leptin is the result of an (unsuccessful) attempt to overcome the leptin resistance. In those very rare humans with extreme obesity who have a defective leptin gene (OB), leptin injection does result in dramatic weight loss. In the vast majority of obese individuals, however, the OB gene is intact. In clinical trials, the injection of leptin did not have the weight-reducing effect observed in obese ob/ob mice. Clearly, most cases of human obesity involve one or more factors in addition to leptin.
Adiponectin Acts through AMPK to Increase Insulin Sensitivity
Adiponectin is a peptide hormone produced almost exclusively in adipose tissue, an adipokine that sensitizes other organs to the effects of insulin. Adiponectin circulates in the blood and powerfully affects the metabolism of fatty acids and carbohydrates in liver and muscle. It increases the uptake of fatty acids from the blood by myocytes and the rate at which fatty acids undergo β oxidation in muscle. It also blocks fatty acid synthesis and gluconeogenesis in hepatocytes, and stimulates glucose uptake and catabolism in muscle and liver.
These effects of adiponectin are indirect and not fully understood, but the AMP-activated protein kinase (AMPK) mediates many of them. Acting through its GPCR, adiponectin triggers phosphorylation and activation of AMPK. Recall that AMPK is activated by factors that signal the need to shift metabolism toward energy generation and away from energy-requiring biosynthesis (Fig. 23-33; see also p. 503). When activated, AMPK profoundly affects the metabolism of individual cells and, through its actions in the brain, the metabolism of the whole animal.
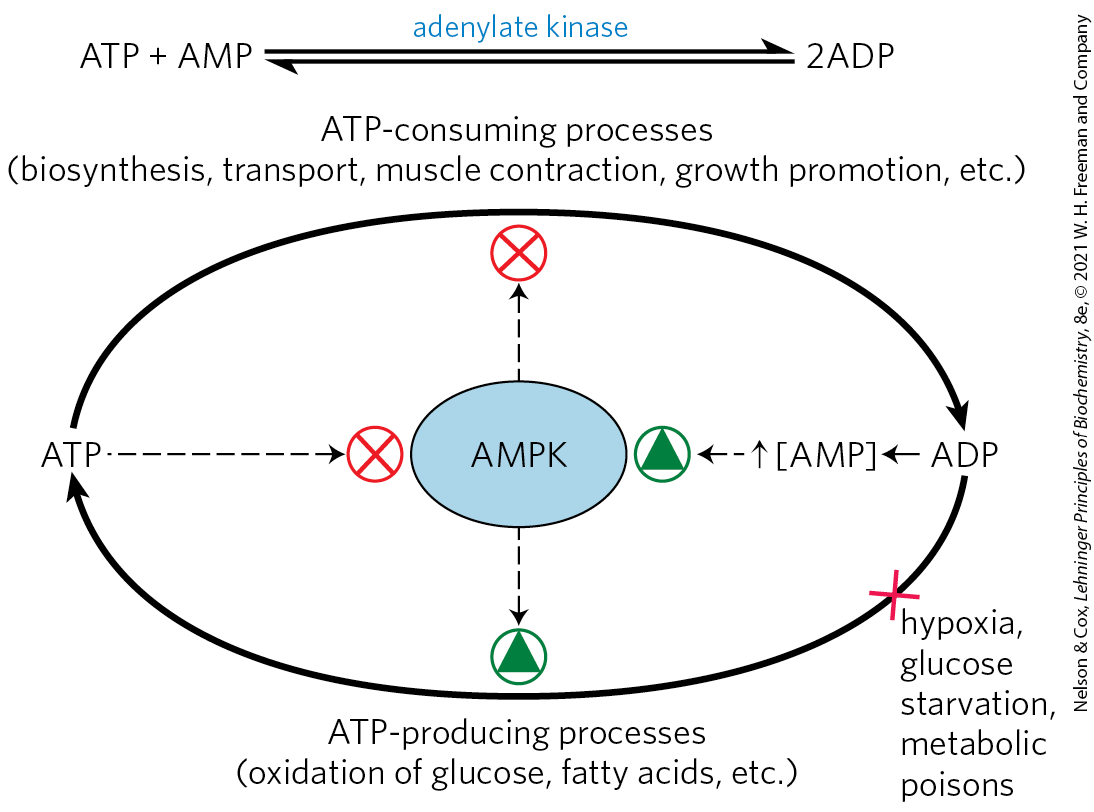
FIGURE 23-33 The role of AMP-activated protein kinase (AMPK) in maintaining energy homeostasis. ADP produced by synthetic reactions is converted to ATP and AMP by adenylate kinase. AMP activates AMPK, which reciprocally regulates ATP-consuming and ATP-generating pathways by phosphorylating key enzymes (see Fig. 23-34). Conditions or agents that inhibit ATP production by catabolic reactions (such as hypoxia, lack of glucose, or metabolic poisons) raise [AMP], activate AMPK, and stimulate catabolism. Cellular or organismal activities that consume ATP (muscle contraction, growth) increase [AMP] and stimulate catabolic reactions to replenish ATP. When [ATP] is high, ATP prevents AMP binding to AMPK, thus lowering AMPK activity and slowing catabolism.
AMPK Coordinates Catabolism and Anabolism in Response to Metabolic Stress
AMPK has emerged as a central player in the coordination of metabolic pathways, organism activity, and feeding behavior (Fig. 23-34). This heterotrimeric, AMP-activated protein kinase monitors the energy and nutrient status in individual cells and shifts metabolism toward energy generation when necessary to maintain metabolic homeostasis. Furthermore, by responding to a variety of hormone signals, AMPK in the hypothalamus acts to keep the whole organism in energetic balance (Fig. 23-8).
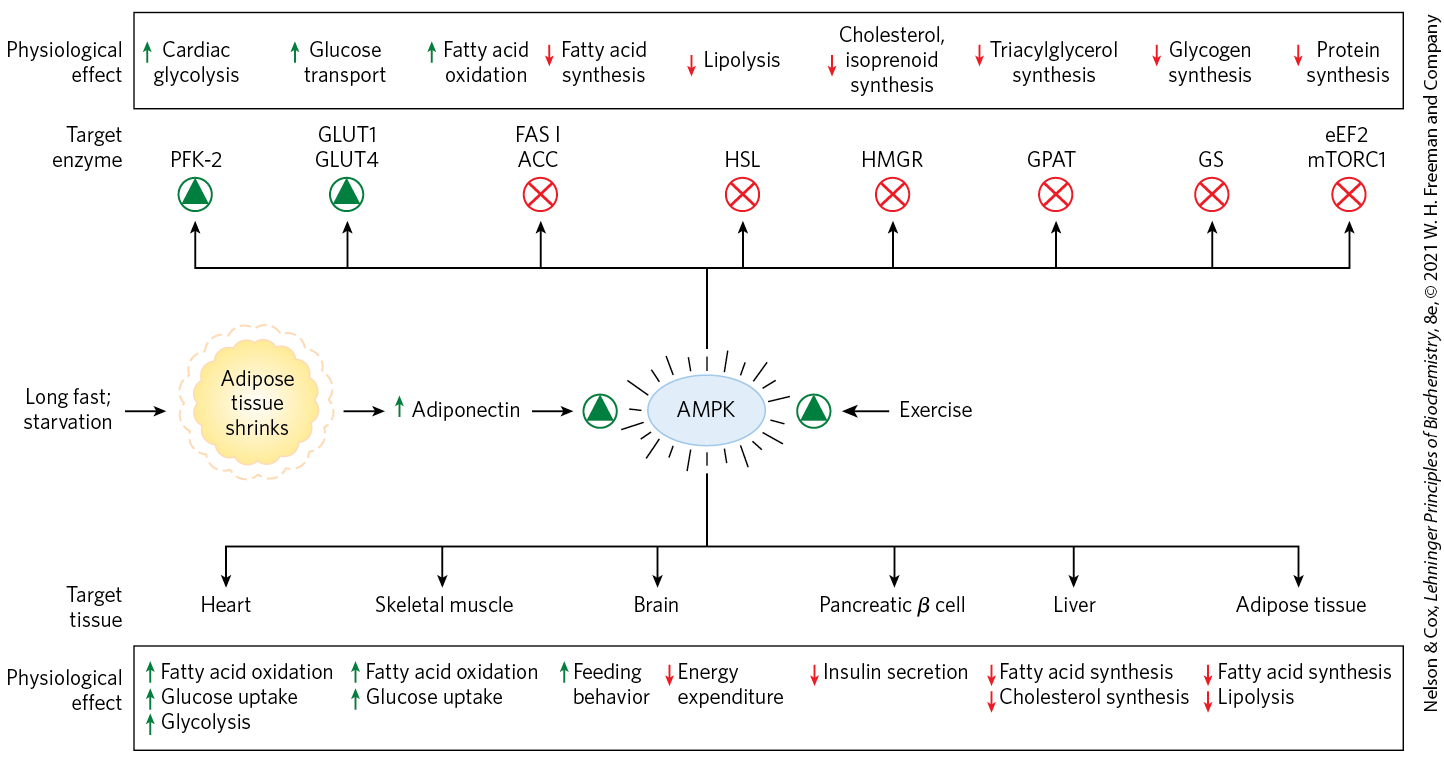
FIGURE 23-34 Formation of adiponectin and its actions through AMPK. Extended fasting or starvation decreases triacylglycerol reserves in adipose tissue, which triggers adiponectin production and release from adipocytes. Adiponectin acts through its plasma membrane receptors in various cell types and organs to inhibit energy-consuming processes and stimulate energy-producing processes. It acts in the brain to stimulate feeding behavior and inhibit energy-consuming physical activity, and in brown fat to inhibit thermogenesis. Adiponectin exerts some of its metabolic effects by activating AMPK, which regulates (by phosphorylation) specific enzymes in key metabolic processes. PFK-2, phosphofructokinase-2; GLUT1 and GLUT4, glucose transporters; FAS I, fatty acid synthase I; ACC, acetyl-CoA carboxylase; HSL, hormone-sensitive lipase; HMGR, HMG-CoA reductase; GPAT, an acyl transferase; GS, glycogen synthase; eEF2, eukaryotic elongation factor 2 (required for protein synthesis; see Chapter 27); and mTORC1, mammalian target of rapamycin complex 1 (a protein kinase complex that regulates protein synthesis on the basis of nutrient availability). Exercise, through conversion of ATP to ADP and AMP, also stimulates AMPK.
AMPK monitors the energy status of a cell through its response to increased [AMP]/[ATP]. Many of the energy-consuming reactions in cells convert ATP to ADP or AMP. Adenylate kinase catalyzes the reaction , so [AMP] is a sensitive measure of the cell’s energy status. AMPK is allosterically activated by AMP binding, and ATP prevents AMP binding, so the enzyme is activated when the cell is energetically depleted (high [AMP]) and inactivated when energy is plentiful (high [ATP], and high [ATP]/[AMP]). AMPK responds to the energetic needs of the whole organism through a second mode of regulation. The enzyme is activated 100-fold by phosphorylation of by liver kinase B1 (LKB1), which is itself subject to regulation by upstream components, including adiponectin. When activated by phosphorylation and AMP binding, AMPK phosphorylates specific enzymes in metabolic pathways that are crucial to energy homeostasis (Fig. 23-34).
When AMPK senses depletion of ATP in an individual cell, lipid synthesis is inhibited and use of lipid as fuel is stimulated. One enzyme regulated by AMPK in the liver and in white adipose tissue is acetyl-CoA carboxylase, which produces malonyl-CoA, the first intermediate committed to fatty acid synthesis. Malonyl-CoA is a powerful inhibitor of the enzyme carnitine acyltransferase 1, which starts the process of β oxidation by transporting fatty acids into the mitochondrion (see Fig. 17-6). By phosphorylating and inactivating acetyl-CoA carboxylase, AMPK inhibits fatty acid synthesis while relieving inhibition (by malonyl-CoA) of β oxidation (see Fig. 17-13). Cholesterol synthesis, a heavy energy consumer, is also inhibited by AMPK, which phosphorylates and inactivates HMG-CoA reductase, an enzyme central to sterol synthesis (see Fig. 21-34). Similarly, AMPK inhibits fatty acid synthase and acyl transferase, effectively blocking the synthesis of TAGs. In addition to its effects on lipid metabolism, AMPK inhibits the synthesis of glycogen and of protein (see Fig. 23-35). Inadequate supplies of oxygen (hypoxia) or blood glucose (hypoglycemia) are among the stressors that trigger AMPK activation.
In the hypothalamus, AMPK is positioned to receive a variety of signals from throughout the body (Fig. 23-8). Ghrelin and adiponectin signal “empty stomach” and “fat tissue depleted,” and, like low blood glucose, they elicit hypothalamic signals, mediated by AMPK, that stimulate feeding and inhibit energy-requiring biosynthetic processes. Leptin, as we have seen, brings to the brain the signal “adipose tissue is full,” slowing catabolic processes and favoring growth and biosynthesis.
At the cytoplasmic surface of lysosomes, AMPK interacts with a second central regulator of cellular activity, the protein kinase mTOR. This enzyme, at the center of an enormous complex, mTORC1, gauges whether sufficient nutrients and low molecular weight substrates are available to support cell growth and proliferation. Together, the two protein kinases, AMPK and mTOR, control major aspects of a cell’s activity and fate.
The mTORC1 Pathway Coordinates Cell Growth with the Supply of Nutrients and Energy
The highly conserved Ser/Thr kinase mTOR forms a complex, mTORC1, with a scaffold protein, raptor, and other regulatory proteins. mTORC1 is recruited to the cytosolic surface of the lysosome through raptor by another complex, Ragulator, and a number of associated Rag G proteins that sense amino acid sufficiency in the cell. The Ragulator-Rag complex is tethered to the lysosomal membrane by covalently linked lipids. When mTORC1 is docked to the Ragulator-Rag complex, it is in contact with another G protein, Rheb (Fig. 23-35). This massive complex integrates signals from inside and outside of the lysosome about the energy status of the cell, the availability of critical amino acids needed for protein synthesis, and the presence of growth factors. When these signals indicate that the cell has what it needs to grow, GTP-bound Rheb activates the protein kinase activity of mTOR, which then phosphorylates many different proteins required for transcription, increased ribosome synthesis, and expression of genes encoding the enzymes of lipid synthesis and mitochondrial proliferation (Fig. 23-36). Because the lysosome is where the cell recycles defective or unneeded components, or extracellular substances brought into the cell through phagocytosis, it is, in effect, the warehouse of parts for cellular construction. The mTORC1-Ragulator-Rag complex is the supply chain manager determining whether the assembly line can operate.
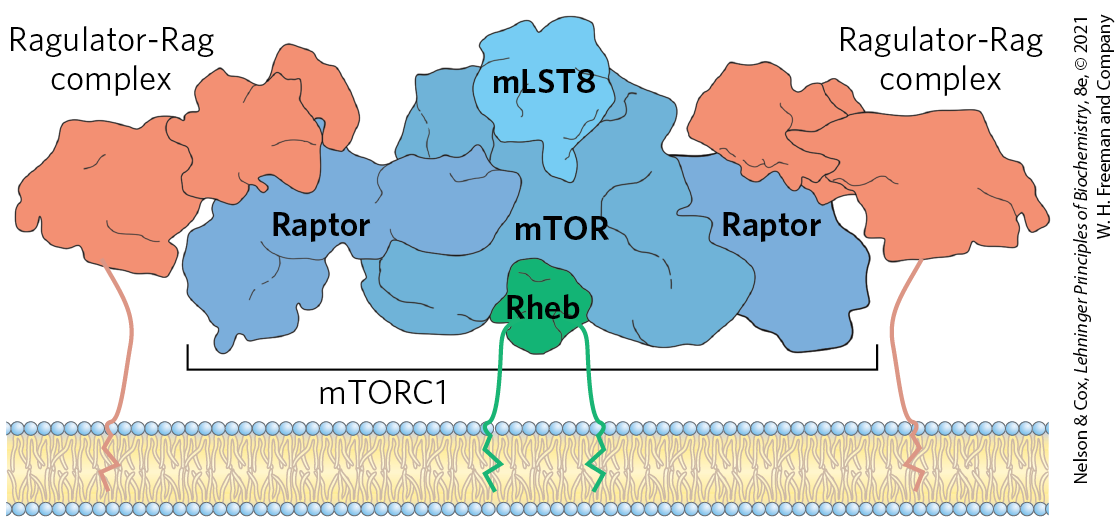
FIGURE 23-35 The mTORC1-Ragulator-Rag complex on the lysosomal surface. This model integrates the structures of mTORC1 and Ragulator-Rag, separately determined by cryo-EM. Raptor acts as a scaffold protein organizing the assembly, which puts the mTOR protein kinase in contact with the G protein Rheb for activation. [Information from J. H. Park et al., Trends Biochem. Sci. 45:367, 2019, Fig. 1; K. B. Rogala et al., Science 366:468, 2019, Fig. 5A.]
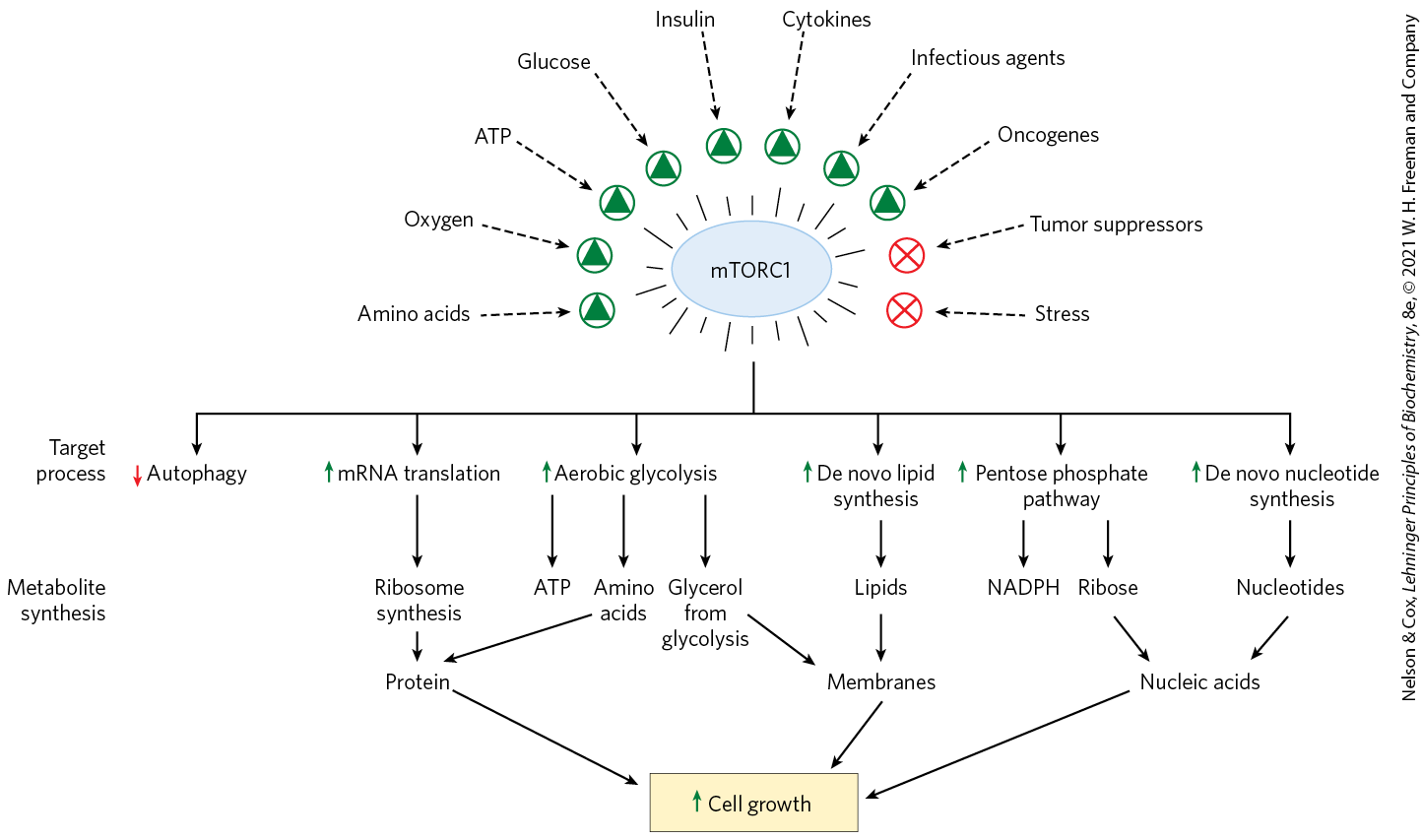
FIGURE 23-36 A summary of mTORC1 activation signals and the cellular processes that active mTORC1 stimulates. The Ser/Thr protein kinase of mTORC1 is activated by the G protein Rheb, reflecting the integration of many signals that indicate that the cell is prepared for growth. By phosphorylating key target proteins, mTORC1 activates energy (ATP and NADPH) production for biosynthesis and stimulates the synthesis of proteins and lipids, allowing cell growth and proliferation.
Fasting results in inactivation of mTORC1 by AMPK, leading to increased breakdown of protein and glycogen in the liver and muscle and mobilization of TAGs in adipose tissue. Chronic activation of mTORC1 by overeating results in excess deposition of TAGs in adipose tissue, as well as in liver and muscle, which may contribute to insulin insensitivity and type 2 diabetes. Mutations that produce constantly activated mTORC1 are commonly associated with human cancers.
Diet Regulates the Expression of Genes Central to Maintaining Body Mass
Proteins in a family of ligand-activated transcription factors known as peroxisome proliferator-activated receptors (PPARs) respond to changes in dietary lipid by altering the expression of genes involved in fat and carbohydrate metabolism. (These transcription factors were first recognized for their roles in peroxisome synthesis — thus their names.) Their normal ligands are fatty acids or fatty acid derivatives. PPARγ, PPARα, and PPARδ act in the nucleus by forming heterodimers with another nuclear receptor, RXR, then binding to regulatory regions of DNA near the genes under their control and changing the rate of transcription of those genes (Fig. 23-37).
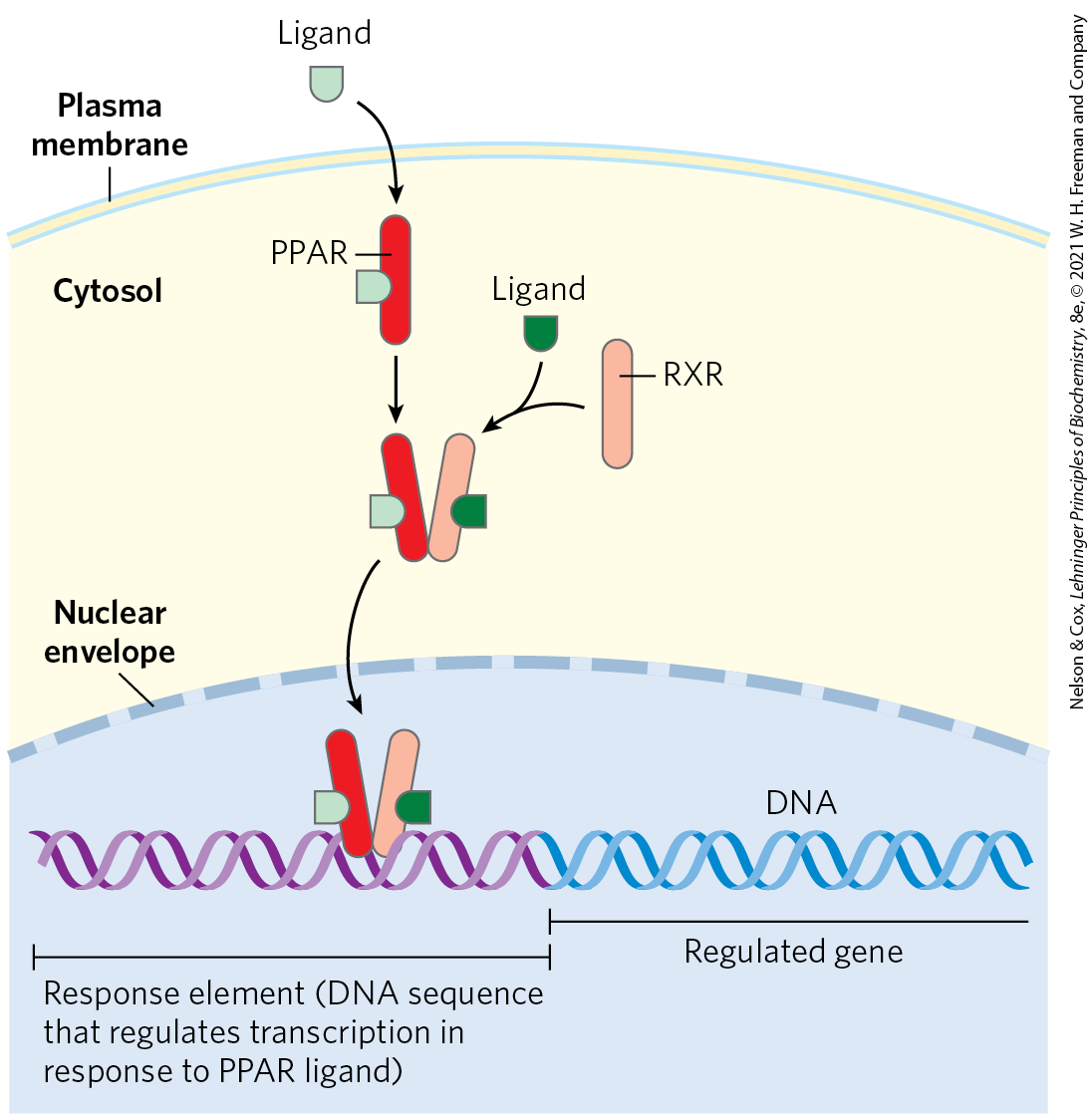
FIGURE 23-37 Mode of action of PPARs. PPARs are transcription factors that, when bound to their cognate ligand, form heterodimers with the nuclear receptor RXR. The dimer binds specific regions of DNA known as response elements, stimulating transcription of genes in those regions. [Information from R. M. Evans et al., Nat. Med. 10:355, 2004, Fig. 3.]
PPARγ turns on genes that act in the differentiation of fibroblasts into adipocytes and genes that encode proteins required for lipid synthesis and storage in adipocytes (Fig. 23-38). PPARγ is activated by the thiazolidinedione drugs that are used to treat type 2 diabetes (discussed below).
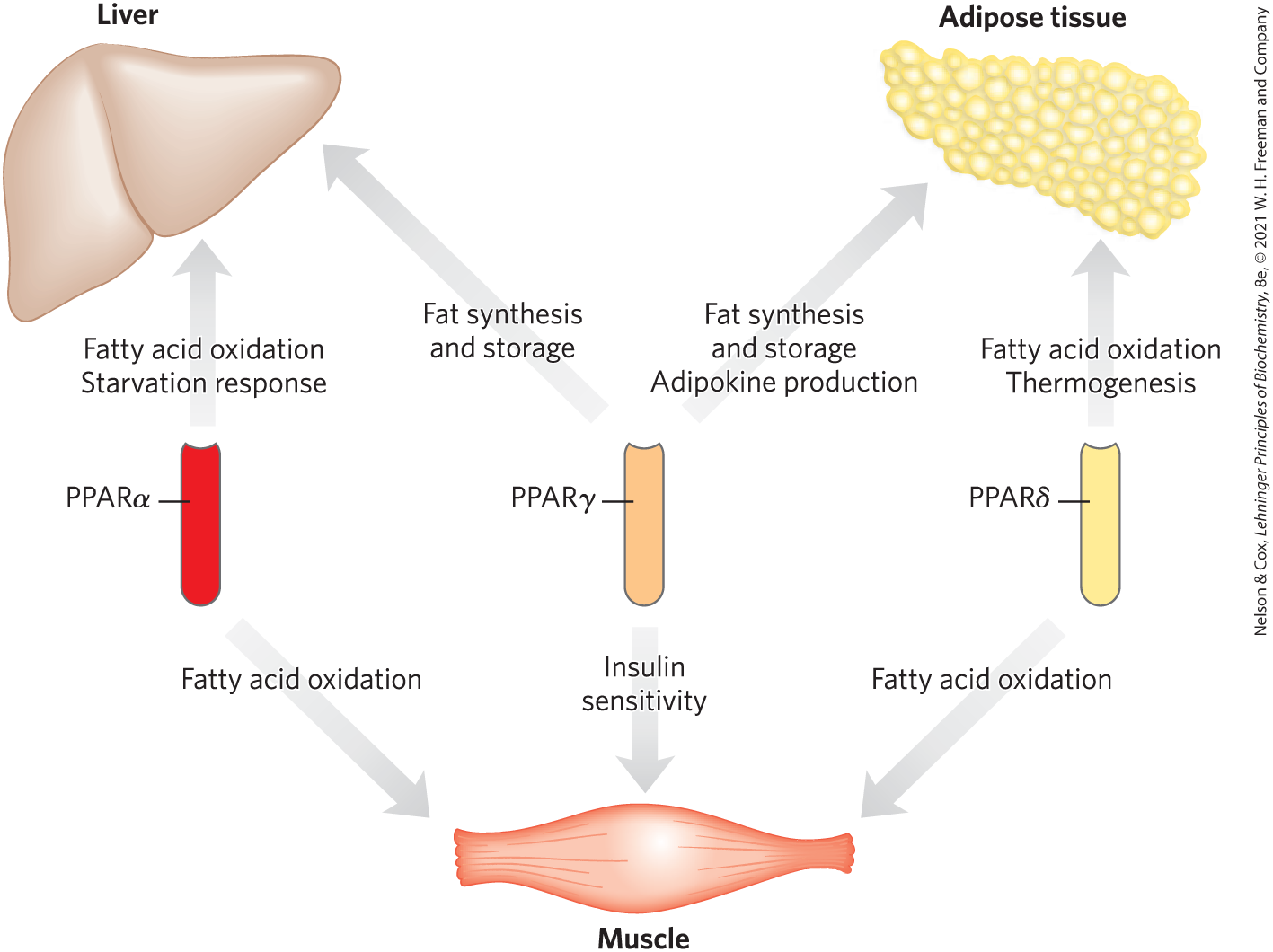
FIGURE 23-38 Metabolic integration by PPARs. The three PPAR isoforms regulate lipid and glucose homeostasis through their coordinated effects on gene expression in liver, muscle, and adipose tissue. PPARα and PPARδ regulate lipid utilization; PPARγ regulates lipid storage and the insulin sensitivity of various tissues.
PPARα is expressed in liver, kidney, heart, skeletal muscle, and brown adipose tissue. The ligands that activate this transcription factor include eicosanoids and free fatty acids. In hepatocytes, PPARα turns on the genes necessary for the uptake and β oxidation of fatty acids and for the formation of ketone bodies during fasting.
PPARδ is a key regulator of fat oxidation, which responds to changes in dietary lipid. PPARδ acts in liver and muscle, stimulating the transcription of at least nine genes encoding proteins for β oxidation and for energy dissipation through uncoupling of mitochondria. By stimulating fatty acid breakdown in uncoupled mitochondria, PPARδ causes fat depletion, weight loss, and thermogenesis.
Short-Term Eating Behavior Is Influenced by Ghrelin, , and Cannabinoids
The peptide hormone ghrelin is produced in cells lining the stomach. It is a powerful appetite stimulant that works on a shorter time scale (between meals) than leptin and insulin. Ghrelin receptors are located in the hypothalamus, affecting appetite, as well as in heart muscle and adipose tissue. Ghrelin acts through a GPCR to generate the second messenger , which mediates the hormone’s action. The concentration of ghrelin in the blood fluctuates strikingly throughout the day, peaking just before a meal and dropping sharply just after a meal (Fig. 23-39). Injection of ghrelin into humans produces immediate sensations of intense hunger. Individuals with Prader-Willi syndrome, whose blood levels of ghrelin are exceptionally high, have an uncontrollable appetite, leading to extreme obesity that often results in death before the age of 30.
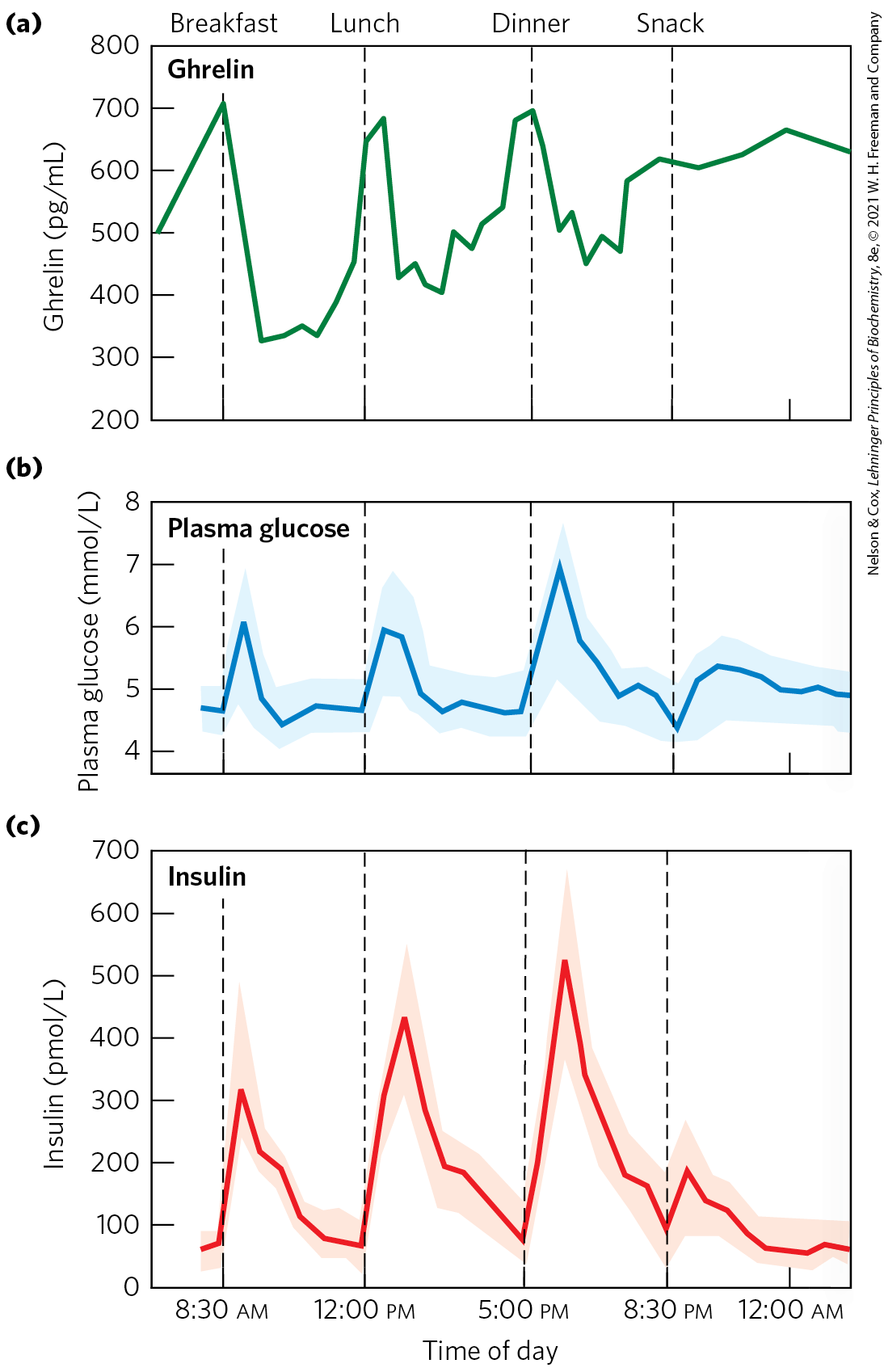
FIGURE 23-39 Variations in blood concentrations of glucose, ghrelin, and insulin relative to meal times. (a) Plasma levels of ghrelin rise sharply just before the usual time for meals (7 am breakfast, 12 noon lunch, 5:30 pm dinner) and drop precipitously just after meals, paralleling subjective feelings of hunger. (b) Plasma glucose rises sharply after a meal, (c) followed immediately by a rise in insulin level in response to the increased blood glucose. [(a, c) Data from D. E. Cummings et al., Diabetes 50:1714, 2001, Fig. 1. (b) Data from M. D. Feher and C. J. Bailey, Br. J. Diabet. Vasc. Dis. 4:39, 2004.]
is a peptide hormone (34 amino acid residues) secreted by endocrine cells in the lining of the small intestine and colon in response to food entering from the stomach. The level of in the blood rises after a meal and remains high for some hours. The hormone is carried in the blood to the arcuate nucleus, where it acts on orexigenic neurons, inhibiting NPY release and reducing hunger (Fig. 23-32). Humans injected with feel little hunger and eat less than normal amounts for about 12 hours.
Endocannabinoids (Fig. 23-40) are eicosanoid lipid messengers that act through specific GPCRs in the brain and peripheral nervous system to increase appetite, heighten the sensory response to food (especially sweet and fatty foods), and elevate mood. When food enters the mouth, neuronal signals travel to the brain, and from there through the vagus nerve to the intestine, which then produces and releases endocannabinoids. The receptors for endocannabinoids control ion channels of sensory neurons, changing their membrane potentials and sending signals to the brain. Palatable food sensed in this way motivates further consumption of that food. The taste of fats (which are particularly high in caloric content) causes cannabinoid release, which effectively triggers further consumption. Well-conserved across vertebrate species, this system probably evolved to maximize the intake of food and to guard against starvation. In mammals, cannabinoid action stimulates an increase in fat mass and inhibits energy loss by motor activity or thermogenesis. Cannabinoid receptors also mediate the psychoactive effects of -tetrahydrocannabinol (Fig. 23-40), the main active ingredient in marijuana, long known for its stimulating effect on appetite.
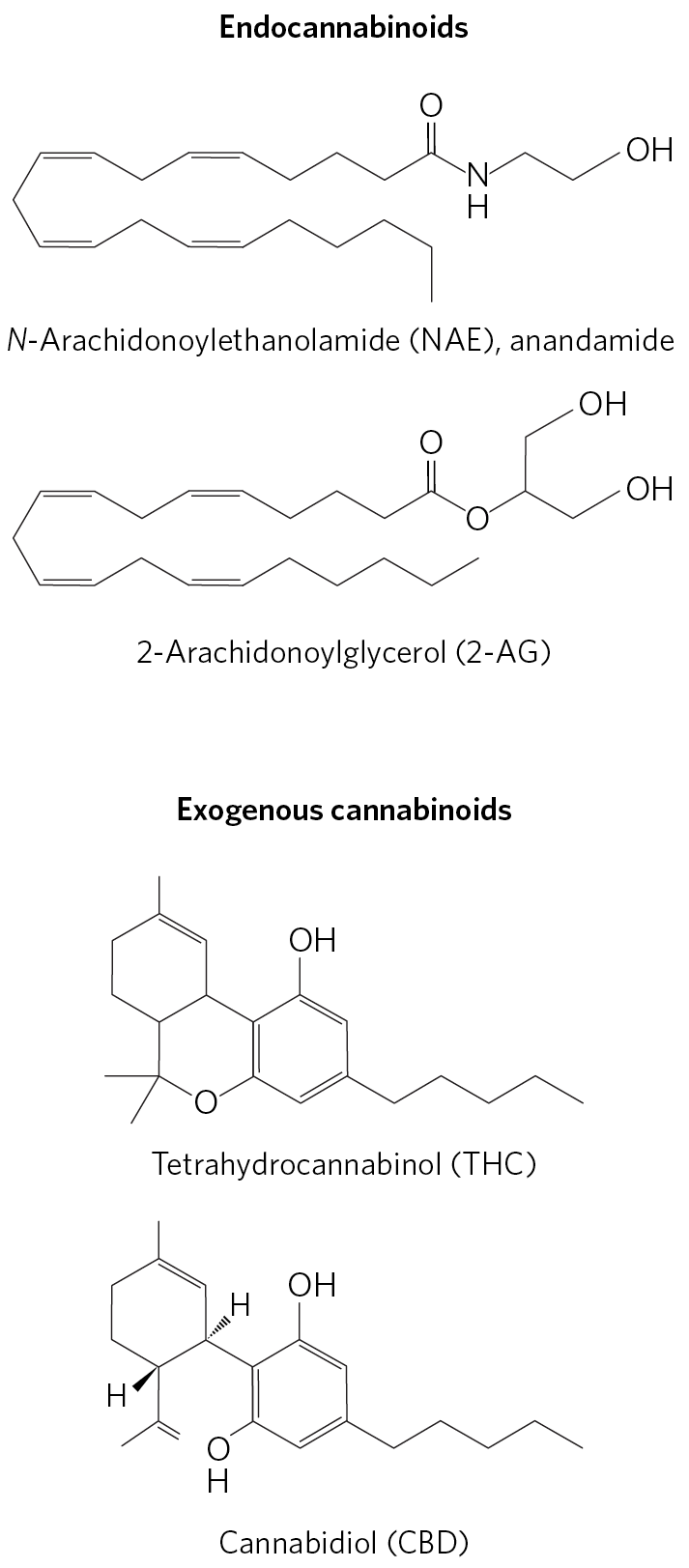
FIGURE 23-40 Cannabinoids. Two endocannabinoids produced by animals, and two products from the cannabis plant. Endocannabinoids carry retrograde signals: secreted by a postsynaptic cell, they diffuse across the synaptic cleft and activate GPCR receptors in the presynaptic cell. The receptors are of two types: in the central nervous system, and in the peripheral nervous system. -Tetrahydrocannabinol (THC), the psychoactive compound in cannabis, is an agonist of both types of cannabinoid receptors in animals; cannabidiol (CBD) does not bind either.
Microbial Symbionts in the Gut Influence Energy Metabolism and Adipogenesis
An adult human is host to about microbial cells that inhabit the gut. These microbes function as a major endocrine organ, producing a variety of metabolites with profound effects on host metabolism, feeding behavior, and body mass. Lean and obese individuals harbor different combinations of microbial symbionts in the gut. Investigation of this observation led to the discovery that gut microbes release fermentation products — the short-chain fatty acids acetate, propionate, butyrate, and lactate — that enter the bloodstream and trigger metabolic changes in adipose tissue (Fig. 23-41). Propionate, for example, drives the expansion of white adipose tissue by acting on GPCRs in the plasma membranes of several cell types, including adipocytes. These receptors trigger differentiation of precursor cells (preadipocytes) into adipocytes and inhibit lipolysis in existing adipocytes, leading to an increase in white adipose tissue mass — that is, obesity. Gut microbes also convert primary bile acids, synthesized in the liver, into the secondary bile acids deoxycholate and lithocholate, which enter the bloodstream and act through GPCRs and steroid receptors to activate beige adipocytes to produce UCP1 and increase energy expenditure.
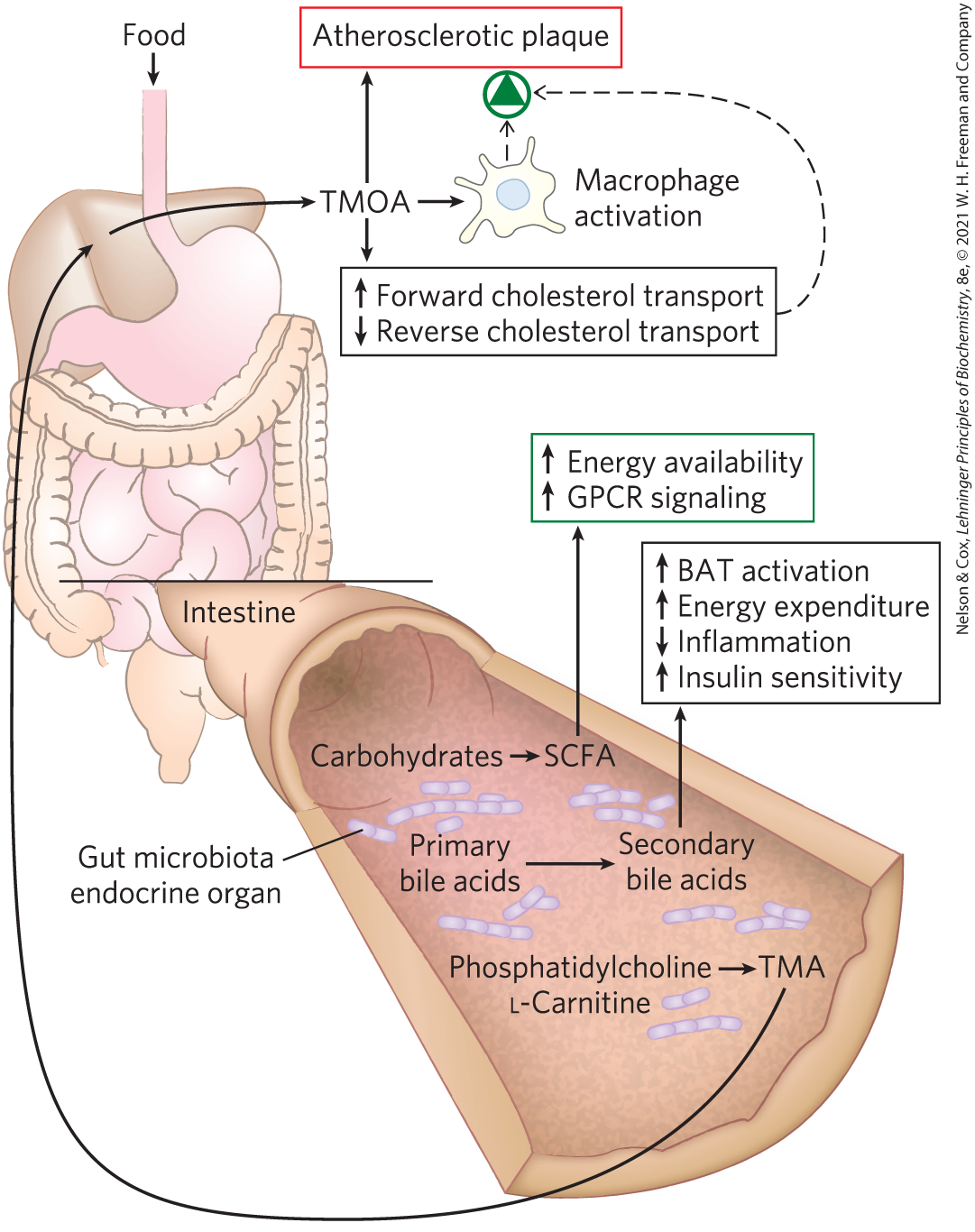
FIGURE 23-41 Effects of gut microbe metabolism on health. The enormous number and diversity of microorganisms (the microbiota) in the colon generate metabolic products that may have significant effects on health, both positive and negative. For example, the metabolism of primary bile acids by microbiota produces secondary products that act through nuclear receptors to stimulate thermogenesis in host brown adipose tissue (BAT) and increase both energy consumption and insulin sensitivity, while reducing inflammation. Metabolism of undigested carbohydrates by microbiota produces short-chain fatty acids (SCFAs) that signal expansion of the host’s white adipose tissue (WAT), promoting obesity. SCFAs produced by microbiota are also a readily metabolizable energy source for the host. Microbial production of the SCFA propionate prevents lipogenesis in the liver and lowers blood cholesterol, both favorable to health. On the other hand, metabolic conversion of phosphatidylcholine and l-carnitine to trimethylamine (TMA), and its further conversion in liver to trimethylamine-N-oxide (TMAO), results in receptor-mediated changes in cholesterol transport and macrophage activity. The combination of altered sterol transport and increased macrophage activity can lead to formation of atherosclerotic plaque (see Fig. 21-46).
These findings raise the possibility of preventing obesity by altering the makeup of the microbial community in the gut. Weight reduction might be accomplished either by adding, directly to the gut, microbial species (probiotics) that disfavor adipogenesis, or by adding to the diet nutrients (prebiotics) that favor the dominance of probiotic microbes. For example, experiments in mice have shown that fructans, polymers of fructose that are indigestible by animals, favor a specific microbial community. When this combination of microorganisms is present, fat storage in white adipose tissue and liver decreases, and there is none of the decrease in insulin sensitivity that is associated with obesity and lipid deposition in the liver (see below). Investigators have transplanted fecal material from lean mice to fat ones, and found that a new collection of microbes became established in the gut of the recipient animals, and the animals lost weight.
Endocrine cells in the lining of the intestinal tract secrete peptides — the anorexigenic and GLP-1 and the orexigenic ghrelin — that modulate food intake and energy expenditure. Interaction with specific microbes in the gut, or with their fermentation products, may trigger release of these peptides. Understanding how diet and microbial symbionts interact to affect energy metabolism and adipogenesis is an important key to understanding the development of obesity, metabolic syndrome, and type 2 diabetes and is a major challenge for the future.
The exquisitely interconnected system of neuroendocrine controls of food intake and metabolism presumably evolved to protect against starvation and to eliminate counterproductive accumulation of fat (extreme obesity). The difficulty most people face in trying to lose weight testifies to the remarkable effectiveness of these controls.
SUMMARY 23.4 Obesity and the Regulation of Body Mass
- Adipose tissue produces leptin, a hormone that regulates feeding behavior and energy expenditure so as to maintain adequate reserves of fat. Leptin production and release increase with the number and size of adipocytes.
- Leptin acts on receptors in the brain, causing the release of anorexigenic (appetite-suppressing) peptides, including α-MSH, that act in the brain to inhibit eating.
- Adiponectin stimulates fatty acid uptake and oxidation and inhibits fatty acid synthesis. It also sensitizes muscle and liver to insulin. At least some of the actions of adiponectin are mediated by AMPK, which is also activated by metabolic stress (low [AMP]), and by exercise.
- The protein kinase complex mTORC1 gauges the supply of essential amino acids and other metabolites, triggering cell growth if all required nutrients are available. It thus complements AMPK in determining the energetic status of a cell.
- Expression of the enzymes of lipid synthesis is under tight and complex regulation. PPARs are transcription factors that determine the rate of synthesis of many enzymes involved in lipid metabolism and adipocyte differentiation.
- Ghrelin, a hormone produced in the stomach, acts on orexigenic (appetite-stimulating) neurons in the arcuate nucleus to produce hunger before a meal. , a peptide hormone of the intestine, acts at the same site to lessen hunger after a meal. Endocannabinoids signal the availability of sweet or fatty food and stimulate its consumption.
- Microbial symbionts in the gut produce fermentation products and secondary bile acids, which influence release of gut hormones that regulate body mass.
 Adipose tissue produces leptin, a hormone that regulates feeding behavior and energy expenditure so as to maintain adequate reserves of fat. Leptin production and release increase with the number and size of adipocytes.
Adipose tissue produces leptin, a hormone that regulates feeding behavior and energy expenditure so as to maintain adequate reserves of fat. Leptin production and release increase with the number and size of adipocytes.