21.4 Cholesterol, Steroids, and Isoprenoids: Biosynthesis, Regulation, and Transport
Cholesterol is doubtless the most publicized lipid, notorious because of the strong correlation between high levels of cholesterol in the blood and the incidence of human cardiovascular diseases. Less well advertised is cholesterol’s crucial role as a component of cellular membranes and as a precursor of steroid hormones and bile acids. Cholesterol is an essential molecule in many animals, including humans, but is not required in the mammalian diet — all cells can synthesize it from simple precursors.
The structure of this 27-carbon compound suggests a complex biosynthetic pathway, but all of its carbon atoms are provided by a single precursor — acetate. The isoprene units that are the essential intermediates in the pathway from acetate to cholesterol are also precursors to many other natural lipids, and the mechanisms by which isoprene units are polymerized are similar in all these pathways.
We begin with an account of the main steps in the biosynthesis of cholesterol from acetate, and then discuss the transport of cholesterol in the blood, its uptake by cells, the normal regulation of cholesterol synthesis, and its regulation in those with defects in cholesterol uptake or transport. We next consider other cellular components derived from cholesterol, such as bile acids and steroid hormones. Finally, an outline of the biosynthetic pathways to some of the many compounds derived from isoprene units, which share early steps with the pathway to cholesterol, illustrates the extraordinary versatility of isoprenoid condensations in biosynthesis.
Cholesterol Is Made from Acetyl-CoA in Four Stages
Cholesterol, like long-chain fatty acids, is made from acetyl-CoA. But the assembly plan of cholesterol is quite different from that of long-chain fatty acids. In early experiments, animals were fed acetate labeled with in either the methyl carbon or the carboxyl carbon. The pattern of labeling in the cholesterol isolated from the two groups of animals in these tracer experiments (Fig. 21-32) provided the blueprint for working out the enzymatic steps in cholesterol biosynthesis.
FIGURE 21-32 Origin of the carbon atoms of cholesterol. This was deduced from tracer experiments with acetate labeled in the methyl carbon (black) or the carboxyl carbon (red). The individual rings in the fused-ring system are designated A through D.
Synthesis takes place in four stages, as shown in Figure 21-33: condensation of three acetate units to form a six-carbon intermediate, mevalonate; conversion of mevalonate to activated isoprene units; polymerization of six 5-carbon isoprene units to form the 30-carbon linear squalene; and cyclization of squalene to form the four rings of the steroid nucleus, with a further series of changes (oxidations, removal or migration of methyl groups) to produce cholesterol.
FIGURE 21-33 Summary of cholesterol biosynthesis. Isoprene units in squalene are set off by dashed red lines.
Stage Synthesis of Mevalonate from Acetate
The first stage in cholesterol biosynthesis leads to the intermediate mevalonate (Fig. 21-34). Two molecules of acetyl-CoA condense to form acetoacetyl-CoA, which condenses with a third molecule of acetyl-CoA to yield the six-carbon compound β-hydroxy-β-methylglutaryl-CoA (HMG-CoA). These first two reactions are catalyzed by acetyl-CoA acetyl transferase and HMG-CoA synthase, respectively. Both reactions are Claisen condensations, and the standard equilibrium in each case favors degradation to acetyl-CoA. However, in cells, the synthetic reactions are facilitated by the rapid utilization of the product HMG-CoA in subsequent reactions. The cytosolic HMG-CoA synthase in this pathway is distinct from the mitochondrial isozyme that catalyzes HMG-CoA synthesis in ketone body formation (see Fig. 17-16).
FIGURE 21-34 Formation of mevalonate from acetyl-CoA. The origin of C-1 and C-2 of mevalonate from acetyl-CoA is shaded light red.
The third reaction is the committed step: reduction of HMG-CoA to mevalonate, for which two molecules of NADPH each donate two electrons. HMG-CoA reductase, an integral membrane protein of the smooth ER, is the major point of regulation on the pathway to cholesterol, as we shall see.
Stage Conversion of Mevalonate to Two Activated Isoprenes
In the next stage, three phosphate groups are transferred from three ATP molecules to mevalonate (Fig. 21-35). The phosphate attached to the C-3 hydroxyl group of mevalonate in the intermediate 3-phospho-5-pyrophosphomevalonate is a good leaving group; in the next step, both this phosphate and the nearby carboxyl group leave, producing a double bond in the five-carbon product, -isopentenyl pyrophosphate. This is the first of the two activated isoprenes central to cholesterol formation. Isomerization of -isopentenyl pyrophosphate yields the second activated isoprene, dimethylallyl pyrophosphate. Synthesis of isopentenyl pyrophosphate in the cytoplasm of plant cells follows the pathway described here. However, plant chloroplasts and many bacteria use a mevalonate-independent pathway. This alternative pathway does not occur in animals, so it is an attractive target for the development of new antibiotics.
FIGURE 21-35 Conversion of mevalonate to activated isoprene units. Six of these activated units combine to form squalene (see Fig. 21-36). The leaving groups of 3-phospho-5-pyrophosphomevalonate are shaded light red. The bracketed intermediate is hypothetical. Both of these isoprene products are required for the next stage in cholesterol biosynthesis.
Stage Condensation of Six Activated Isoprene Units to Form Squalene
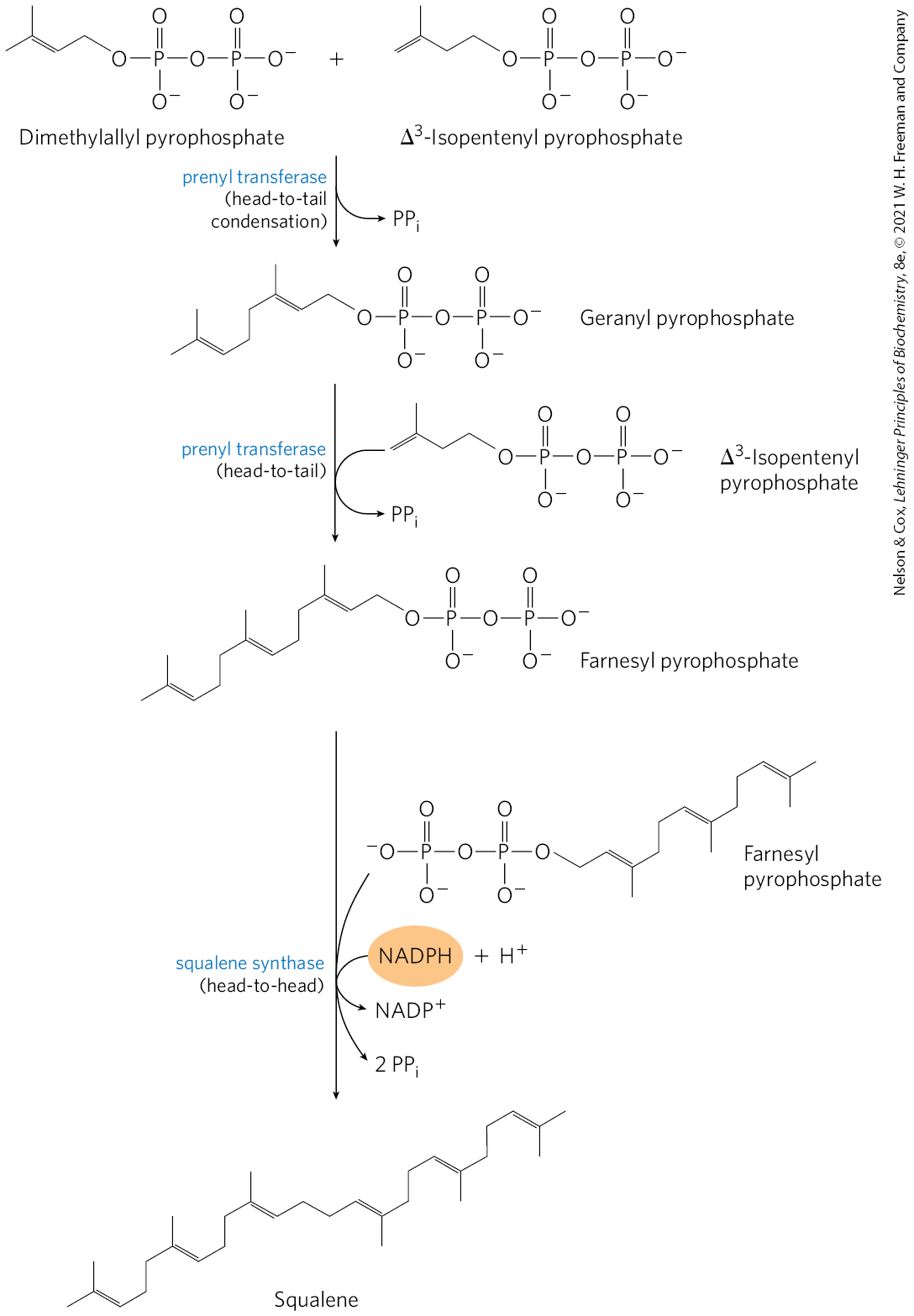
Isopentenyl pyrophosphate and dimethylallyl pyrophosphate now undergo a head-to-tail condensation, in which one pyrophosphate group is displaced and a 10-carbon chain, geranyl pyrophosphate, is formed (Fig. 21-36). (The “head” is the end to which pyrophosphate is joined.) Geranyl pyrophosphate undergoes another head-to-tail condensation with isopentenyl pyrophosphate, yielding the 15-carbon intermediate farnesyl pyrophosphate. Finally, two molecules of farnesyl pyrophosphate join head to head, with the elimination of both pyrophosphate groups, to form squalene. Squalene has 30 carbons: 24 in the main chain and 6 in the form of methyl group branches.
FIGURE 21-36 Formation of squalene. This 30-carbon structure arises through successive condensations of activated isoprene (five-carbon) units.
The common names of these intermediates derive from the sources from which they were first isolated. Geraniol, a component of rose oil, has the aroma of geraniums, and farnesol is an aromatic compound found in flowers of the Farnese acacia tree. Many natural scents of plant origin are synthesized from isoprene units. Squalene was first isolated from the liver of sharks (genus Squalus).
Stage Conversion of Squalene to the Four-Ring Steroid Nucleus
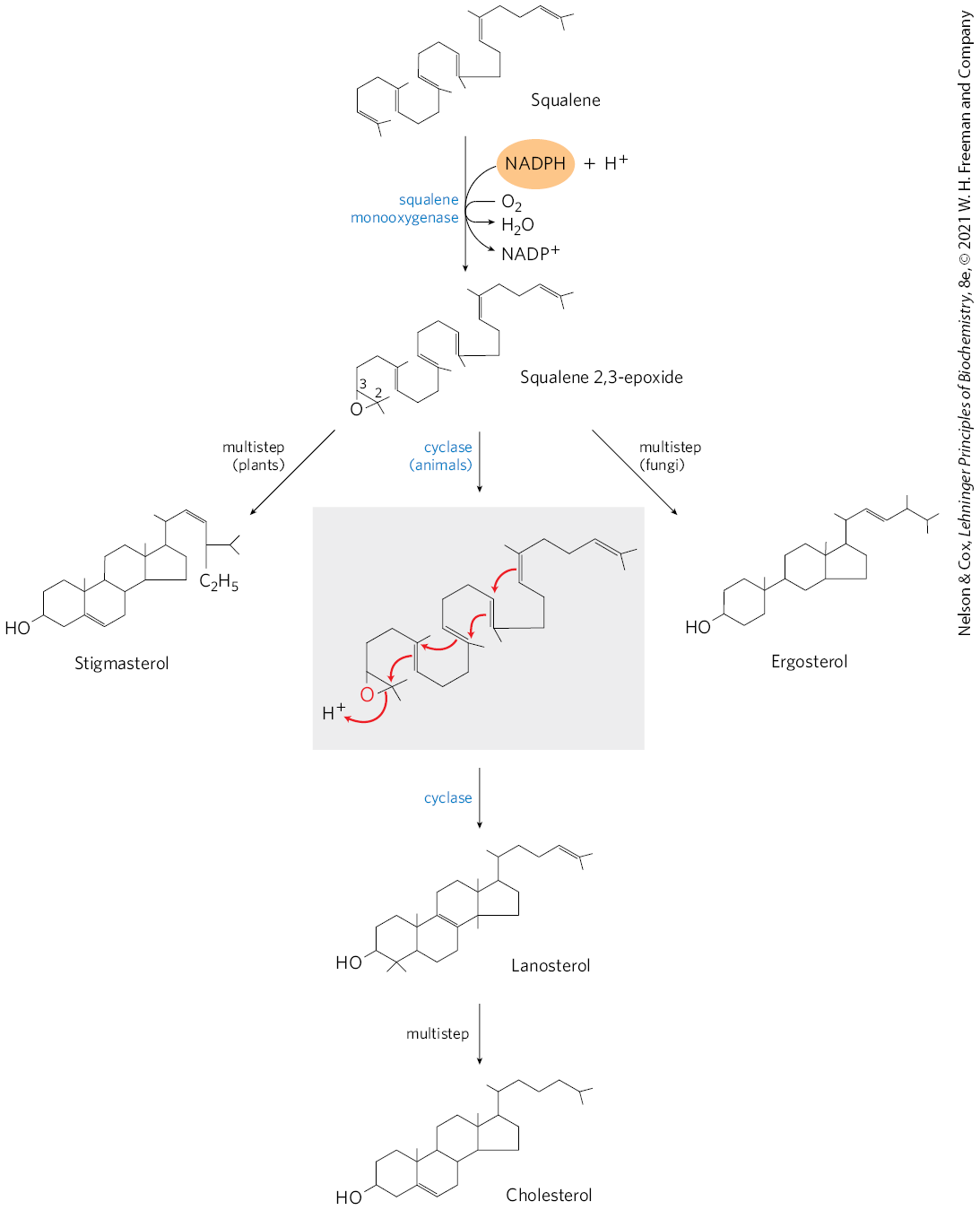
When the squalene molecule is represented as in Figure 21-37, the relationship of its linear structure to the cyclic structure of the sterols becomes apparent. All sterols have the four fused rings that form the steroid nucleus, and all are alcohols, with a hydroxyl group at C-3 — thus the name “sterol.” The action of squalene monooxygenase adds one oxygen atom from to the end of the squalene chain, forming an epoxide. This enzyme is another mixed-function oxidase; NADPH reduces the other oxygen atom of to . The double bonds of the product, squalene 2,3-epoxide, are positioned so that a remarkable concerted reaction can convert the linear squalene epoxide to a cyclic structure. In animal cells, this cyclization results in the formation of lanosterol, which contains the four rings characteristic of the steroid nucleus. Lanosterol is finally converted to cholesterol in a series of about 20 reactions that include the migration of some methyl groups and the removal of others. Elucidation of this extraordinary biosynthetic pathway, one of the most complex known, was accomplished by Konrad Bloch, Feodor Lynen, John Cornforth, and George Popják in the late 1950s.
FIGURE 21-37 Ring closure converts linear squalene to the condensed steroid nucleus. The first step in this sequence is catalyzed by a mixed-function oxygenase, for which the cosubstrate is NADPH. The product is an epoxide, which in the next step is cyclized to the steroid nucleus. The final product of these reactions in animal cells is cholesterol; in other organisms, slightly different sterols are produced, as shown.
Cholesterol is the sterol characteristic of animal cells; plants, fungi, and protists make other, closely related sterols instead. They use the same synthetic pathway as far as squalene 2,3-epoxide, at which point the pathways diverge slightly, yielding other sterols, such as stigmasterol in many plants and ergosterol in fungi (Fig. 21-37).
WORKED EXAMPLE 21-1 Energetic Cost of Squalene Synthesis
What is the energetic cost of the synthesis of squalene from acetyl-CoA, in number of ATPs per molecule of squalene synthesized?
SOLUTION:
In the pathway from acetyl-CoA to squalene, ATP is consumed only in the steps that convert mevalonate to the activated isoprene precursors of squalene. Three ATP molecules are used to create each of the six activated isoprenes required to construct squalene, for a total cost of 18 ATP molecules.
Cholesterol Has Several Fates
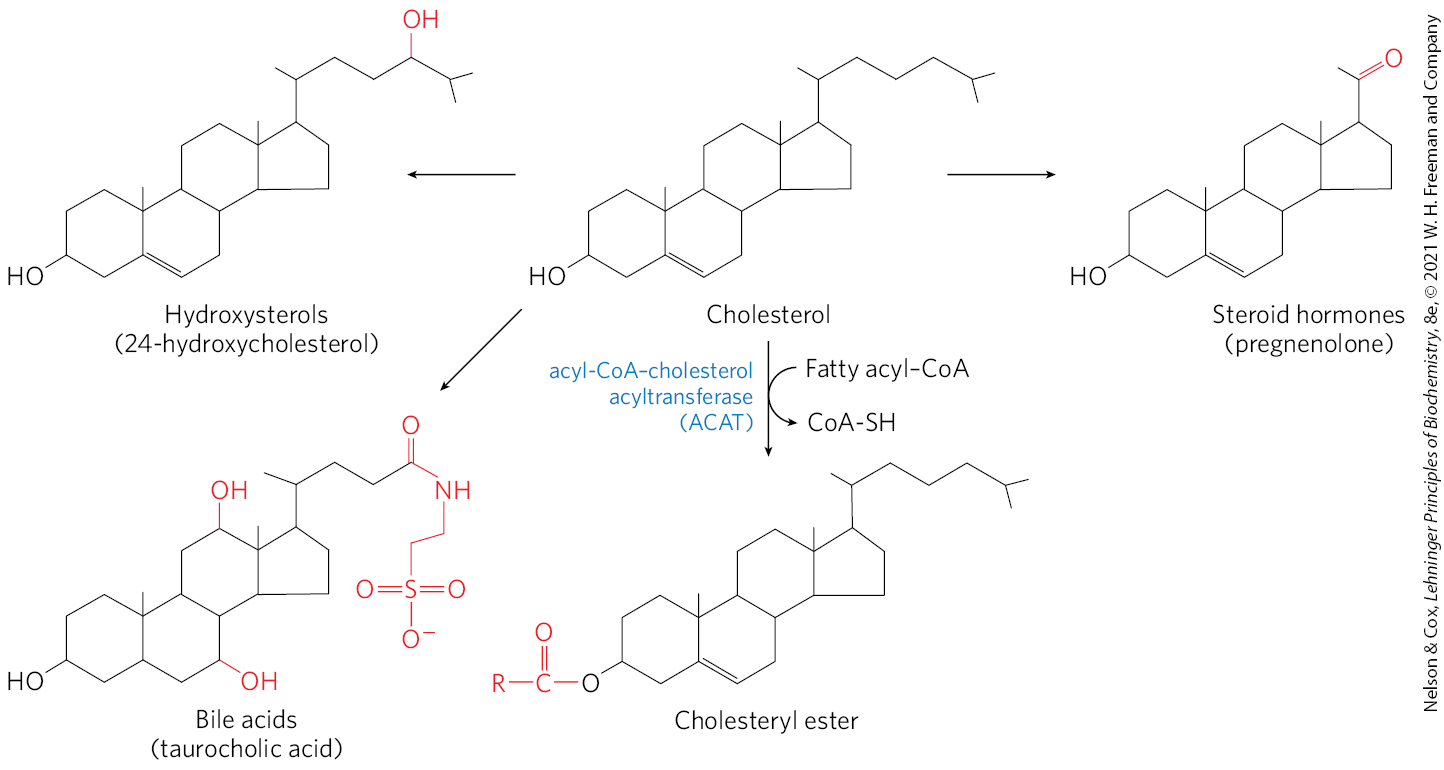
Most of the cholesterol synthesis in vertebrates takes place in the liver. A small fraction of the cholesterol made there is incorporated into the membranes of hepatocytes, but most of it is exported in one of three forms: as bile acids, as biliary cholesterol, or as cholesteryl esters (Fig. 21-38). Small quantities of oxysterols such as 25-hydroxycholesterol are formed in the liver and act as regulators of cholesterol synthesis (see below). In other tissues, cholesterol is converted into steroid hormones (in the adrenal cortex and gonads, for example; see Fig. 10-18) or into vitamin D hormone (in the liver and kidney; see Fig. 10-19). Such hormones are extremely potent biological signals acting through nuclear receptor proteins.
FIGURE 21-38 Metabolic fates of cholesterol. Modifications of the cholesterol structure are shown in red. Esterification converts cholesterol to an even more hydrophobic form for storage and transport; each of the other modifications yields a less hydrophobic product.
Bile acids, one of the three forms of cholesterol exported from the liver, are the principal components of bile, a fluid stored in the gallbladder and excreted into the small intestine to aid in the digestion of fat-containing meals. Bile acids and their salts are relatively hydrophilic cholesterol derivatives that serve as emulsifiers in the intestine, converting large particles of fat into tiny micelles and thereby greatly increasing the surface at which digestive lipases can act (see Fig. 17-1). Bile also contains much smaller amounts of cholesterol (biliary cholesterol). Bile helps remove excess cholesterol from the intestine and facilitates excretion. Dietary fiber can enhance this effect by binding to bile and interfering with bile reabsorption in the intestines, leading to increased bile excretion in the feces. More cholesterol is then used to make bile. The soluble fiber available in oats (oatmeal) and barley is especially effective.
Cholesteryl esters are formed in the liver through the action of acyl-CoA–cholesterol acyltransferase (ACAT). This enzyme catalyzes the transfer of a fatty acid from coenzyme A to the hydroxyl group of cholesterol (Fig. 21-38), converting the cholesterol to a more hydrophobic form that is no longer sufficiently amphipathic to function appropriately in membranes. Cholesteryl esters are transported in secreted lipoprotein particles to other tissues that use cholesterol, or they are stored in the liver in lipid droplets.
Cholesterol and Other Lipids Are Carried on Plasma Lipoproteins
Cholesterol and cholesteryl esters, like triacylglycerols and phospholipids, are essentially insoluble in water, yet they must be moved from the tissue of origin to the tissues in which they will be stored or consumed. They are carried in the blood plasma as plasma lipoproteins, macromolecular complexes of specific carrier proteins, called apolipoproteins, and various combinations of phospholipids, cholesterol, cholesteryl esters, and triacylglycerols.
Apolipoproteins (“apo” designates the protein in its lipid-free form) combine with lipids to form several classes of lipoprotein particles, spherical complexes with hydrophobic lipids in the core and hydrophilic amino acid side chains at the surface (Fig. 21-39a, b). Different combinations of lipids and proteins produce particles of different densities, ranging from chylomicrons to high-density lipoproteins. These particles can be separated by ultracentrifugation (Table 21-1) and visualized by electron microscopy (Fig. 21-39c).
FIGURE 21-39 Lipoproteins. (a) Structure of a chylomicron. Apolipoprotein B-48 (apoB-48) defines the chylomicron. Through the life cycle of a chylomicron other apolipoproteins, including apoC-II, apoC-III, and apoE, become part of the particle, acting as signals in the uptake and metabolism of chylomicron contents. Chylomicrons range from about 100 to 500 nm in diameter. (b) Structure of a low-density lipoprotein (LDL). Apolipoprotein B-100 (apoB-100) is one of the largest single polypeptide chains known, with 4,636 amino acid residues . One particle of LDL contains a core with about 1,500 molecules of cholesteryl esters, surrounded by a shell composed of about 500 more molecules of cholesterol, 800 molecules of phospholipids, and one molecule of apoB-100. (c) Four classes of lipoproteins, visualized in the electron microscope after negative staining. The chylomicrons shown here are 50 to 200 nm in diameter; VLDL, 28 to 70 nm; LDL, 20 to 25 nm; and HDL, 8 to 11 nm. Particle sizes given are those measured for these samples; particle sizes vary considerably in different preparations. For properties of lipoproteins, see Table 21-1. [(b) Data for apoB-100 from A. Johs et al., J. Biol. Chem. 281:19,732, 2006. (c) Robert Hamilton, Jr., PhD.]
TABLE 21-1 Major Classes of Human Plasma Lipoproteins: Some Properties
Composition (wt %)
Lipoprotein
Density (g/mL)
Protein
Phospholipids
Free cholesterol
Cholesteryl esters
Triacylglycerols
Chylomicrons
2
9
1
3
85
VLDL
0.95–1.006
10
18
7
12
50
LDL
1.006–1.063
23
20
8
37
10
HDL
1.063–1.210
55
24
2
15
4
Data from D. Kritchevsky, Nutr. Int. 2:290, 1986.
Each class of lipoprotein has a specific function, determined by its point of synthesis, lipid composition, and apolipoprotein content. At least 10 distinct apolipoproteins are found in the lipoproteins of human plasma (Table 21-2), distinguishable by their size, their reactions with specific antibodies, and their characteristic distribution in the lipoprotein classes. These protein components act as signals, targeting lipoproteins to specific tissues or activating enzymes that act on the lipoproteins. Figure 21-40 provides an overview of the formation and transport of the lipoproteins in mammals. The numbered steps in the following discussion refer to this figure.
TABLE 21-2 Apolipoproteins of the Human Plasma Lipoproteins
Apolipoprotein
Polypeptide molecular weight
Lipoprotein association
Function (if known)
ApoA-I
28,100
HDL
Activates LCAT; interacts with ABC transporter
ApoA-II
17,400
HDL
Inhibits LCAT
ApoA-IV
44,500
Chylomicrons, HDL
Activates LCAT; cholesterol transport/clearance
ApoB-48
242,000
Chylomicrons
Cholesterol transport/clearance
ApoB-100
512,000
VLDL, LDL
Binds to LDL receptor
ApoC-I
7,000
VLDL, HDL
ApoC-II
9,000
Chylomicrons, VLDL, HDL
Activates lipoprotein lipase
ApoC-III
9,000
Chylomicrons, VLDL, HDL
Inhibits lipoprotein lipase
ApoD
32,500
HDL
ApoE
34,200
Chylomicrons, VLDL, HDL
Triggers clearance of VLDL and chylomicron remnants
ApoH
50,000
Possibly VLDL, binds phospholipids such as cardiolipin
Roles in coagulation, lipid metabolism, apoptosis, inflammation
Information from D. E. Vance and J. E. Vance (eds), Biochemistry of Lipids and Membranes, 5th edn, Elsevier Science Publishing, 2008.
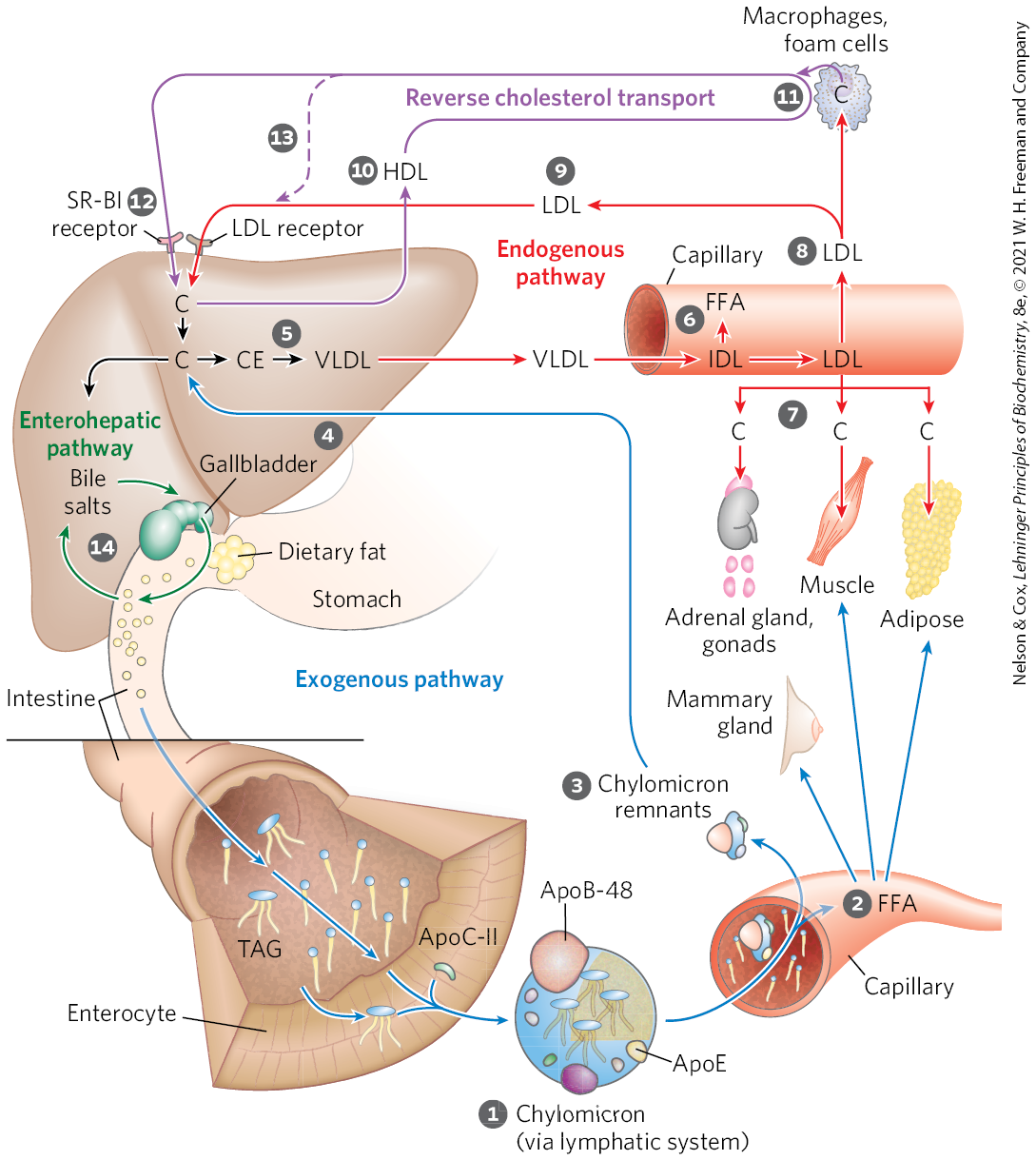
FIGURE 21-40 Lipoproteins and lipid transport. Lipids are transported in the bloodstream as lipoproteins, which exist as several variants that have different functions, different protein and lipid compositions (see Tables 21-1 and 21-2), and thus different densities. Numbered steps are described in the text. In the exogenous pathway (blue arrows), dietary lipids are packaged into chylomicrons; fatty acids from triacylglycerol (TAG) are released by lipoprotein lipase to adipose and muscle tissues, during transport through capillaries. Chylomicron remnants (containing largely protein and cholesterol) are taken up by the liver. Bile salts produced in the liver aid in dispersing dietary fats and are then reabsorbed in the enterohepatic pathway (green arrows). In the endogenous pathway (red arrows), lipids synthesized or packaged in the liver are delivered to peripheral tissues by VLDL. Extraction of lipid from VLDL (along with loss of some apolipoproteins) gradually converts some of it to LDL, which delivers cholesterol to extrahepatic tissues or returns to the liver. Excess cholesterol in extrahepatic tissues is transported back to the liver as HDL in reverse cholesterol transport (purple arrows). C represents cholesterol; CE, cholesteryl ester.
Chylomicrons, discussed in Chapter 17 in connection with the movement of dietary triacylglycerols from the intestine to other tissues, are the first of four classes of lipoproteins we will discuss. These are the largest of the lipoproteins and the least dense, containing a high proportion of triacylglycerols. Chylomicrons are synthesized from dietary fats in the ER of enterocytes, epithelial cells that line the small intestine. The chylomicrons then move through the lymphatic system and enter the bloodstream via the left subclavian vein. The apolipoproteins of chylomicrons include apoA-IV, apoB-48 (unique to this class of lipoproteins), apoE, apoC-II, and apoC-III (Table 21-2). ApoC-II activates lipoprotein lipase in the capillaries of adipose, heart, skeletal muscle, and lactating mammary tissues, allowing the release of free fatty acids (FFA) to these tissues. Chylomicrons thus carry dietary fatty acids to tissues where they will be consumed or stored as fuel. The remnants of chylomicrons, depleted of most of their triacylglycerols but still containing cholesterol, apoE, and apoB-48, move through the bloodstream to the liver. Receptors in the liver bind to the apoE in the chylomicron remnants and mediate uptake of these remnants by endocytosis. In the liver, the remnants release their cholesterol and are degraded in lysosomes. This pathway from dietary cholesterol to the liver is the exogenous pathway (blue arrows in Fig. 21-40).
Very-low-density lipoprotein (VLDL) is the second of the four classes. When the diet contains more fatty acids and cholesterol than are needed immediately as fuel or as precursors to other molecules, they are converted to triacylglycerols or cholesteryl esters in the liver and packaged with specific apolipoproteins into VLDL. Excess carbohydrate in the diet can also be converted to triacylglycerols in the liver and exported as VLDL. In addition to triacylglycerols and cholesteryl esters, VLDL contains apoB-100, apoC-I, apoC-II, apoC-III, and apoE (Table 21-2). VLDL is transported in the blood from the liver to muscle and adipose tissue. In the capillaries of these tissues, apoC-II activates lipoprotein lipase, which catalyzes the release of free fatty acids from triacylglycerols in the VLDL. Adipocytes take up these fatty acids, reconvert them to triacylglycerols, and store the products in intracellular lipid droplets; myocytes, in contrast, primarily oxidize the fatty acids to supply energy. When the insulin level is high (after a meal), VLDL serves primarily to convey lipids from the diet to adipose tissue for storage. In the fasting state between meals, the fatty acids used to produce VLDL in the liver originate primarily from adipose tissue, and the principal VLDL target is myocytes of the heart and skeletal muscle.
Low-density lipoprotein (LDL), the third class of lipoprotein, is formed when triacylglycerol loss converts some VLDL to VLDL remnants, also called intermediate-density lipoprotein (IDL). Further removal of triacylglycerol from IDL (remnants) produces LDL. Rich in cholesterol and cholesteryl esters, and containing apoB-100 as its major apolipoprotein, LDL carries cholesterol to extrahepatic tissues such as muscle, adrenal glands, and adipose tissue. These tissues have plasma membrane LDL receptors that recognize apoB-100 and mediate uptake of cholesterol and cholesteryl esters. LDL also delivers cholesterol to macrophages, sometimes converting them into foam cells (see Fig. 21-46). LDL not taken up by peripheral tissues and cells returns to the liver and is taken up via LDL receptors in the hepatocyte plasma membrane. Cholesterol that enters hepatocytes by this path may be incorporated into membranes, converted to bile acids, or reesterified by ACAT (Fig. 21-38) for storage within cytosolic lipid droplets. This pathway, from VLDL formation in the liver to LDL return to the liver, is the endogenous pathway of cholesterol metabolism and transport (red arrows in Fig. 21-40). Accumulation of excess intracellular cholesterol is prevented by reducing the rate of cholesterol synthesis when sufficient cholesterol is available from LDL in the blood. Regulatory mechanisms to accomplish this are described below.
HDL Carries Out Reverse Cholesterol Transport
High-density lipoprotein (HDL), the fourth major lipoprotein in mammals, originates in the liver and small intestine as small, protein-rich particles that contain relatively little cholesterol and no cholesteryl esters (Fig. 21-40). HDLs contain primarily apoA-I and other apolipoproteins (Table 21-2). They also contain the enzyme lecithin-cholesterol acyltransferase (LCAT), which catalyzes the formation of cholesteryl esters from lecithin (phosphatidylcholine) and cholesterol (Fig. 21-41). LCAT on the surface of nascent (newly forming) HDL particles converts the cholesterol and phosphatidylcholine of chylomicron and VLDL remnants encountered in the bloodstream to cholesteryl esters, which begin to form a core, transforming the disk-shaped nascent HDL to a mature, spherical HDL particle. Nascent HDL can also pick up cholesterol from cholesterol-rich extrahepatic cells (including macrophages and foam cells, formed from macrophages; see below). Mature HDL then returns to the liver, where the cholesterol is unloaded via the scavenger receptor SR-BI. Some of the cholesteryl esters in HDL can also be transferred to LDL by the cholesteryl ester transfer protein. The HDL circuit is reverse cholesterol transport (purple arrows in Fig. 21-40). Much of this cholesterol is converted to bile salts by enzymes sequestered in hepatic peroxisomes; the bile salts are stored in the gallbladder and excreted into the intestine when a meal is ingested. Bile salts are reabsorbed by the liver and recirculate through the gallbladder in this enterohepatic circulation (green arrows in Fig. 21-40).
FIGURE 21-41 Reaction catalyzed by lecithin-cholesterol acyltransferase (LCAT). This enzyme is present on the surface of HDL and is stimulated by the HDL component apoA-I. Cholesteryl esters accumulate within nascent HDLs, converting them to mature HDLs.
The unloading of sterols via SR-BI receptors in liver and other tissues does not occur by endocytosis, the mechanism used for LDL uptake. Instead, when HDL binds to SR-BI receptors in the plasma membranes of hepatocytes or steroidogenic tissues such as the adrenal gland, these receptors mediate partial and selective transfer of cholesterol and other lipids in HDL into the cell. Depleted HDL then dissociates to recirculate in the bloodstream and extract more lipids from remnants of chylomicrons and VLDL, and from cells overloaded with cholesterol, as described below.
Cholesteryl Esters Enter Cells by Receptor-Mediated Endocytosis
Each LDL particle in the bloodstream contains apoB-100, which is recognized by LDL receptors present in the plasma membranes of cells that need to take up cholesterol. Figure 21-42 shows such a cell. LDL receptors are synthesized in the ER and transported to the plasma membrane, after modification in the Golgi complex. At the plasma membrane, they are available to bind apoB-100. Binding of LDL to an LDL receptor initiates endocytosis, which conveys the LDL and its receptor into the cell within an endosome. The receptor-containing portions of the endosome membrane bud off and are returned to the cell surface, to function again in LDL uptake. The endosome fuses with a lysosome, which contains enzymes that hydrolyze the cholesteryl esters, releasing cholesterol and fatty acids into the cytosol. The apoB-100 protein is also degraded to amino acids that are released to the cytosol. ApoB-100 is also present in VLDL, but its receptor-binding domain is not available for binding to the LDL receptor; conversion of VLDL to LDL exposes the receptor-binding domain of apoB-100.
FIGURE 21-42 Uptake of cholesterol by receptor-mediated endocytosis.
This pathway for the transport of cholesterol in blood and its receptor-mediated endocytosis by target tissues was elucidated by Michael Brown and Joseph Goldstein. They discovered that individuals with the genetic disease familial hypercholesterolemia (FH) have mutations in the LDL receptor that prevent the normal uptake of LDL by liver and peripheral tissues. The result of defective LDL uptake is very high blood levels of LDL (and of the cholesterol it carries). Individuals with FH have a greatly increased probability of developing atherosclerosis, a disease of the cardiovascular system in which blood vessels are occluded by cholesterol-rich plaques (see Fig. 21-46).
Michael Brown and Joseph Goldstein
Niemann-Pick type-C (NPC) disease is an inherited defect in lipid storage. In this disorder, cholesterol is not transported out of the lysosomes and instead accumulates in lysosomes of liver, brain, and lung, bringing about early death. NPC is the result of a mutation in either of two genes, NPC1 and NPC2, essential to moving cholesterol out of the lysosome and into the cytosol, where it can be further metabolized. NPC1 encodes a transmembrane lysosomal protein, and NPC2 encodes a soluble protein. These proteins act in tandem to transfer cholesterol out of the lysosome and into the cytosol for further processing or metabolism.
Cholesterol Synthesis and Transport Are Regulated at Several Levels
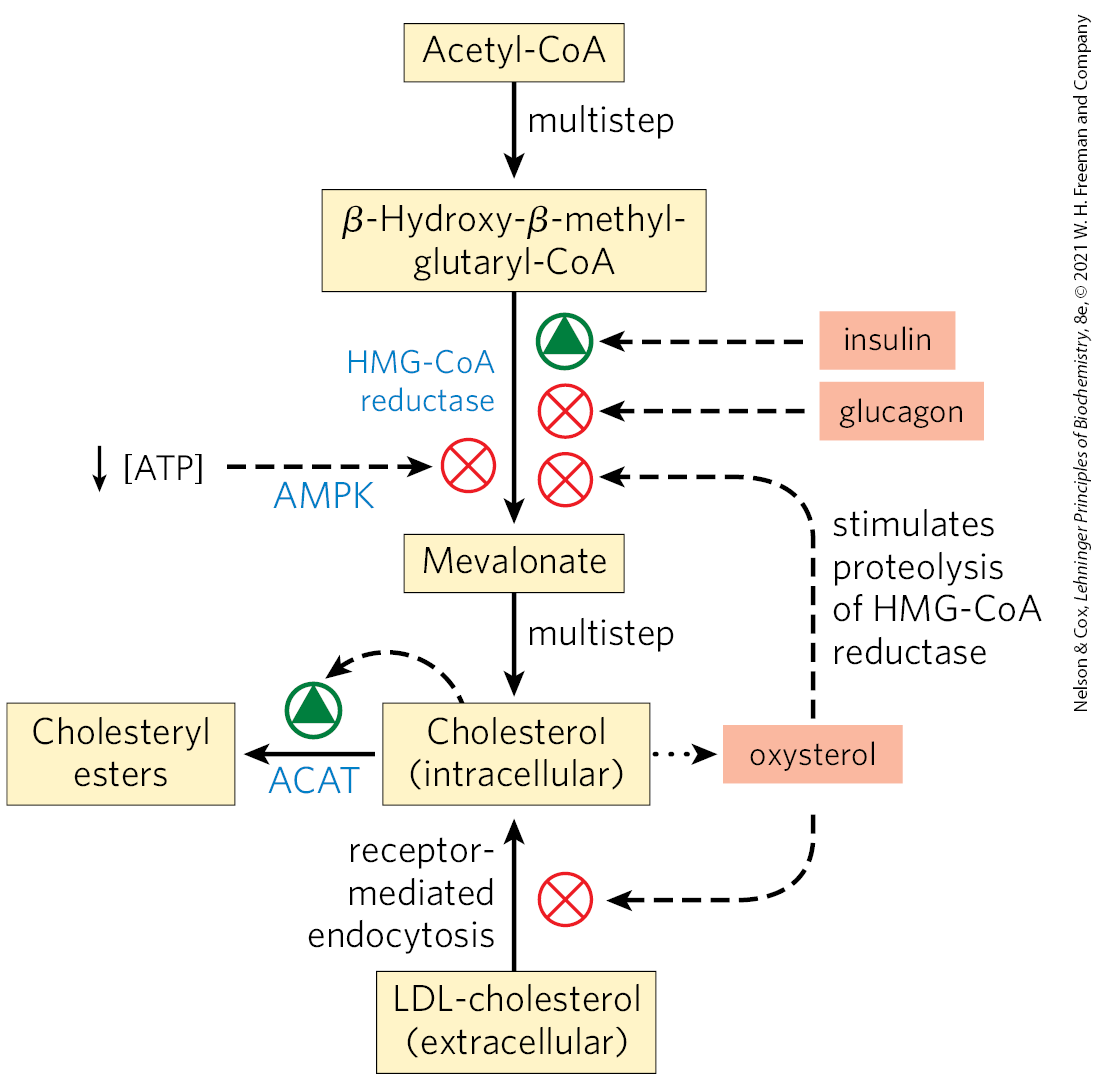
Cholesterol synthesis is a complex and energy-expensive process. Excess cholesterol cannot be catabolized for use as fuel and must be excreted. Therefore, it is clearly advantageous to an organism to regulate the biosynthesis of cholesterol to complement dietary intake. In mammals, cholesterol production is regulated by intracellular cholesterol concentration, by the supply of ATP, and by the hormones glucagon and insulin. The committed step in the pathway to cholesterol (and a major site of regulation) is the conversion of HMG-CoA to mevalonate (Fig. 21-34), the reaction catalyzed by HMG-CoA reductase.
Short-term (minute-to-minute) regulation of the activity of existing HMG-CoA reductase is accomplished by reversible covalent alteration: phosphorylation by the AMP-dependent protein kinase (AMPK), which senses high AMP concentration (indicating low ATP concentration). Thus, when ATP levels drop, the synthesis of cholesterol slows, and catabolic pathways for the generation of ATP are stimulated (Fig. 21-43). Hormones that mediate global regulation of lipid and carbohydrate metabolism also act on HMG-CoA reductase; glucagon stimulates its phosphorylation (inactivation), and insulin promotes dephosphorylation, activating the enzyme and favoring cholesterol synthesis. These covalent regulatory mechanisms are probably not as important, quantitatively, as the mechanisms that affect the synthesis and degradation of the enzyme.
FIGURE 21-43 Regulation of cholesterol formation balances synthesis with dietary uptake and energy state. Insulin promotes dephosphorylation (activation) of HMG-CoA reductase; glucagon promotes its phosphorylation (inactivation); and the AMP-dependent protein kinase AMPK, when activated by low [ATP] relative to [AMP], phosphorylates and inactivates HMG-CoA reductase. Oxysterol metabolites of cholesterol (for example, 24(S)-hydroxycholesterol) stimulate proteolysis of HMG-CoA reductase.
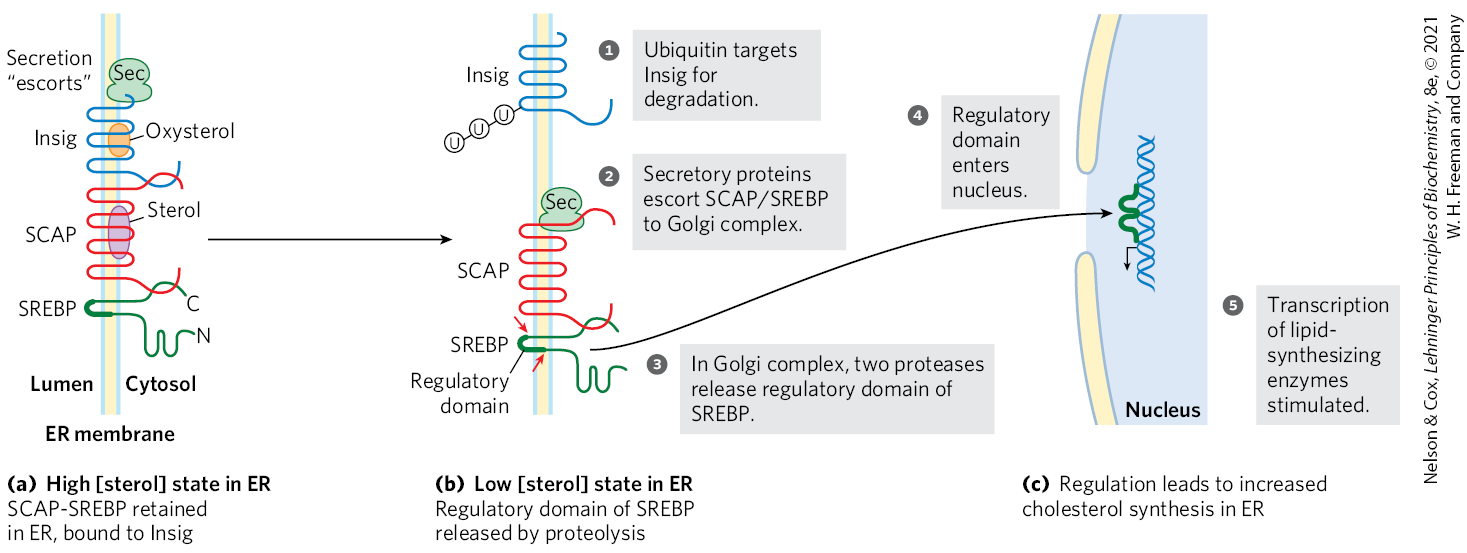
In the longer term, the number of molecules of HMG-CoA reductase is increased or decreased in response to cellular concentrations of cholesterol. Regulation of HMG-CoA reductase synthesis by cholesterol is mediated by an elegant system of transcriptional regulation of the HMG-CoA gene (Fig. 21-44). This gene, along with more than 20 other genes encoding enzymes that mediate the uptake and synthesis of cholesterol and unsaturated fatty acids, is controlled by a small family of proteins called sterol regulatory element-binding proteins (SREBPs). When newly synthesized, these proteins are embedded in the ER. Only the soluble regulatory domain fragment of an SREBP functions as a transcription activator, through mechanisms discussed in Chapter 28. When cholesterol and oxysterol levels are high, SREBPs are held in the ER in a complex with another protein called SREBP cleavage-activating protein (SCAP), which in turn is anchored in the ER membrane by its interaction with a third membrane protein, Insig (insulin-induced gene protein) (Fig. 21-44a). SCAP and Insig act as sterol sensors. When sterol levels are high, the Insig-SCAP-SREBP complex is retained in the ER membrane. When the level of sterols in the cell declines (Fig. 21-44b), the SCAP-SREBP complex is escorted by secretory proteins to the Golgi complex. There, two proteolytic cleavages of SREBP release a regulatory fragment, which enters the nucleus and activates transcription of its target genes, including those for HMG-CoA reductase, the LDL receptor protein, and other proteins needed for lipid synthesis. When sterol levels increase sufficiently, the proteolytic release of SREBP amino-terminal domains is again blocked, and proteasome degradation of the existing active domains results in rapid shutdown of the gene targets.
FIGURE 21-44 Regulation of cholesterol synthesis by SREBP. Sterol regulatory element-binding proteins (SREBPs) are embedded in the ER when first synthesized, in a complex with the protein SREBP cleavage-activating protein (SCAP), which is in turn bound to Insig. (N and C represent the amino and carboxyl termini of the proteins.) (a) Under normal circumstances (high [sterol]), SCAP and Insig are bound to SREBPs and the SREBPs are inactive. (b) When sterol levels decline, sterol-binding sites on Insig and SCAP are unoccupied, and Insig is targeted for degradation by the attachment of several ubiquitin molecules. The remaining SCAP-SREBP complex migrates to the Golgi complex, and SREBP is cleaved (arrows) to produce a regulatory domain fragment. (c) This domain acts in the nucleus to increase the transcription of sterol-regulated genes. [Information from R. Raghow et al., Trends Endocrinol. Metab. 19:65, 2008, Fig. 2.]
The level of HMG-CoA reductase is also regulated by proteolytic degradation of the enzyme itself. High levels of cellular cholesterol are sensed by Insig, which triggers attachment of ubiquitin molecules to HMG-CoA reductase, leading to its degradation by proteasomes.
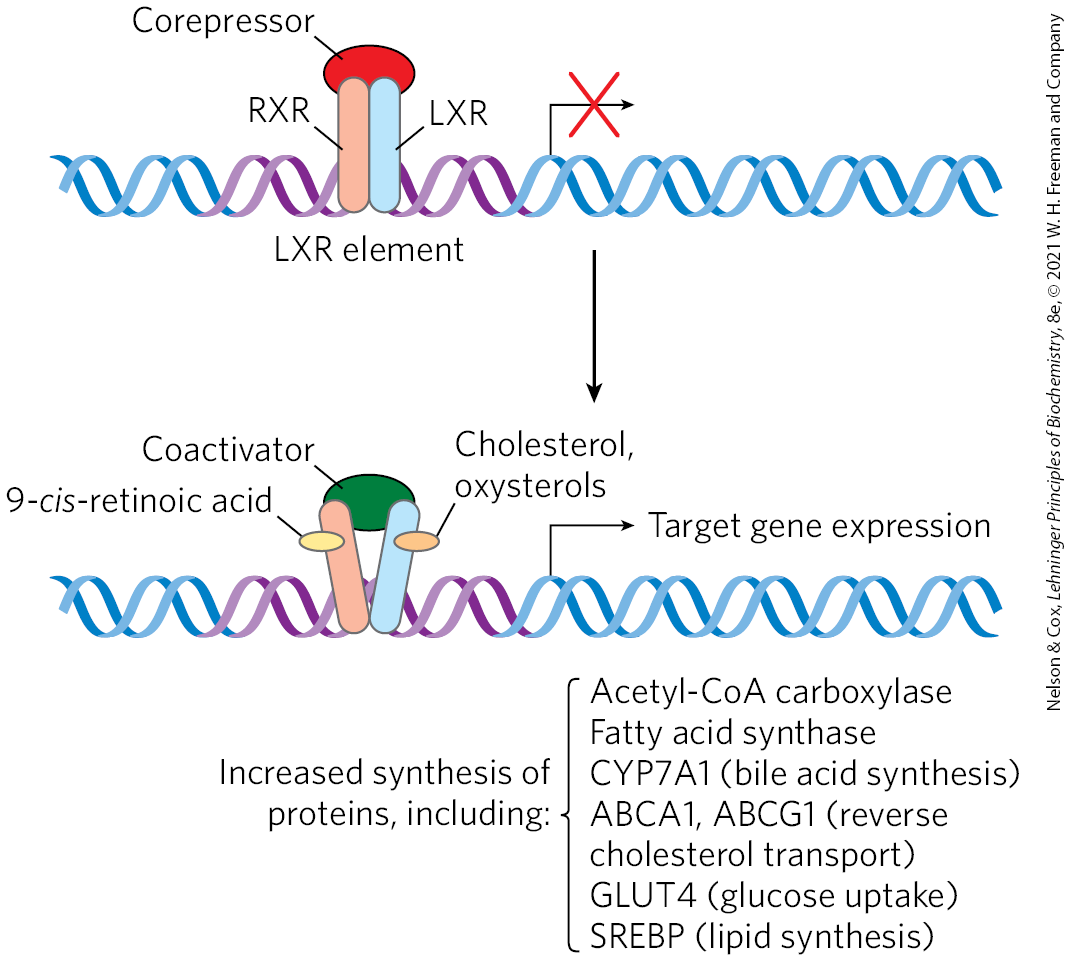
Liver X receptor (LXR) is a nuclear transcription factor activated by oxysterol ligands (reflecting high cholesterol levels), which integrates the metabolism of fatty acids, sterols, and glucose. is expressed primarily in liver, adipose tissue, and macrophages; is present in all tissues. When bound to an oxysterol ligand, LXRs form heterodimers with a second type of nuclear receptor, the retinoid X receptors (RXR), and the LXR-RXR dimer activates transcription from a set of genes (Fig. 21-45), including those for acetyl-CoA carboxylase, the first enzyme in fatty acid synthesis; fatty acid synthase; the cytochrome P-450 enzyme CYP7A1, required for sterol conversion to bile acids; apoproteins that participate in cholesterol transport (apoC-I, apoC-II, apoD, and apoE); the ATP-binding cassette (ABC) transporters ABCA1 and ABCG1, required for reverse cholesterol transport (see below); GLUT4, the insulin-stimulated glucose transporter of muscle and adipose tissue; and an SREBP called SREBP1C. The transcriptional regulators LXR and SREBP therefore work together to achieve and maintain cholesterol homeostasis; SREBPs are activated by low levels of cellular cholesterol, and LXRs are activated by high cholesterol levels.
FIGURE 21-45 Action of RXR-LXR dimer on expression of genes for lipid and glucose metabolism. When their ligands are absent, RXR and LXR associate with a corepressor protein, preventing transcription of the genes associated with the LXR element (LXRE). When their respective ligands are present — 9-cis-retinoic acid for RXR, cholesterol or oxysterols for LXR — the dimer dissociates from the corepressor, then associates with a coactivator protein. This complex binds to the LXR element and turns on expression of the associated genes. Regulation of gene expression is a topic discussed in more detail in Chapter 28. [Information from A. C. Calkin and P. Tontonoz, Nat. Rev. Mol. Cell Biol. 13:213, 2012, Fig. 1.]
Regulation by LXRs is complemented by the activity of farnesoid X receptor (FXR), which also forms a heterodimer with RXR, with an effect that is often reciprocal to that of LXR-RXR. Although farnesol is a ligand for this receptor, FXR responds primarily to bile acids. High levels of bile acids can be toxic. FXR, expressed mainly in the intestine, liver, kidney, and adrenal glands, provides essential control of bile acid levels by increasing or decreasing expression of multiple genes. FXR represses many of the genes that are activated by LXR.
Finally, two other regulatory mechanisms influence cellular cholesterol level: (1) high cellular concentrations of cholesterol activate ACAT, which increases esterification of cholesterol for storage, and (2) high cellular cholesterol levels diminish (via SREBP) transcription of the gene that encodes the LDL receptor, reducing production of the receptor and thus the uptake of cholesterol from the blood.
Dysregulation of Cholesterol Metabolism Can Lead to Cardiovascular Disease
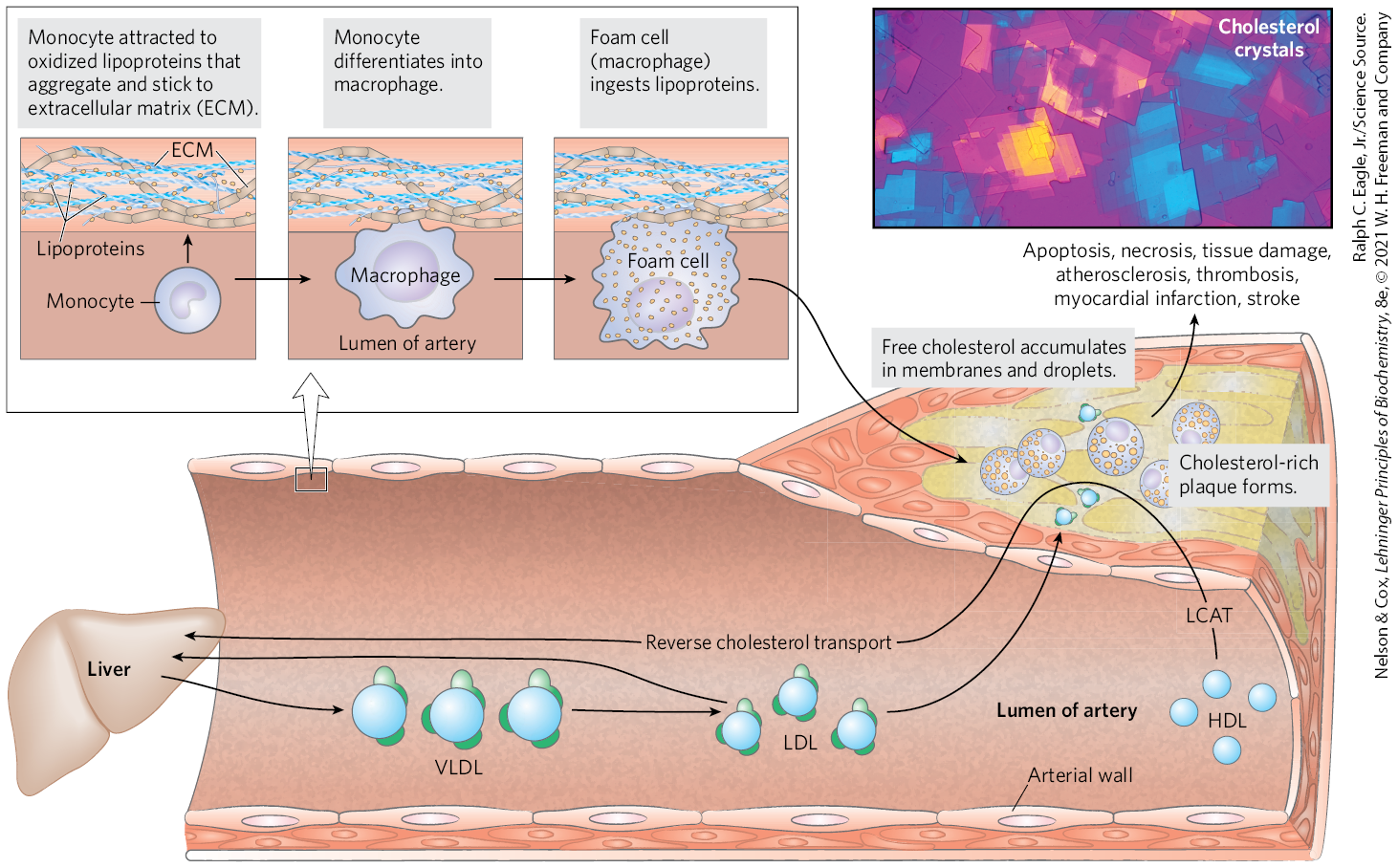
As noted earlier, cholesterol cannot be catabolized by animal cells. Excess cholesterol can be removed only by excretion or by conversion to bile salts. When the sum of cholesterol synthesized and cholesterol obtained in the diet exceeds the amount required for the synthesis of membranes, bile salts, and steroids, pathological accumulations of cholesterol (plaques) can obstruct blood vessels, a condition called atherosclerosis. Heart failure due to occluded coronary arteries is a leading cause of death in industrialized societies. Atherosclerosis is linked to high levels of cholesterol in the blood, and particularly to high levels of LDL-cholesterol (“bad cholesterol”); there is a negative correlation between HDL (“good cholesterol”) levels and arterial disease. Plaque formation in blood vessels is initiated when LDL containing partially oxidized fatty acyl groups adheres to and accumulates in the extracellular matrix of epithelial cells lining arteries (Fig. 21-46). Immune cells (monocytes) are attracted to regions with such LDL accumulations, and they differentiate into macrophages, which take up the oxidized LDL and the cholesterol they contain. Macrophages cannot limit their uptake of sterols, and with increasing accumulation of cholesteryl esters and free cholesterol, the macrophages become foam cells (they appear foamy when viewed under the microscope). As excess free cholesterol accumulates in foam cells and their membranes, the cells undergo apoptosis. Over long periods of time, arteries become progressively occluded as plaques consisting of extracellular matrix material, scar tissue formed from smooth muscle tissue, and foam cell remnants gradually become larger. Within the cholesterol-rich plaques, cholesterol can crystallize (Fig. 21-46, inset). Occasionally, a plaque breaks loose from the site of its formation and is carried through the blood to a narrowed region of an artery in the brain or the heart, causing a stroke or a heart attack. Cholesterol crystals that break loose from plaques can cause vascular injury.
FIGURE 21-46 Formation of atherosclerotic plaques. Excess lipid derived from the diet is deposited on arterial walls, a process facilitated by the conversion of monocytes to foam cells and incorporation of foam cells into growing plaques. Some of this deposition is countered by HDL and reverse cholesterol transport. The LCAT reaction is shown in Figure 21-41. The inset shows a polarized light micrograph of cholesterol crystals. [Ralph C. Eagle, Jr./Science Source.]
In familial hypercholesterolemia, blood levels of cholesterol are extremely high and severe atherosclerosis develops in childhood. Affected individuals have a defective LDL receptor and lack receptor-mediated uptake of cholesterol carried by LDL. Consequently, cholesterol is not cleared from the blood; it accumulates in foam cells and contributes to the formation of atherosclerotic plaques. Endogenous cholesterol synthesis continues despite the excessive cholesterol in the blood, because extracellular cholesterol cannot enter cells to regulate intracellular synthesis (Fig. 21-44). Drugs in a class called statins, some isolated from natural sources and some synthesized industrially, are used to treat patients with elevated serum cholesterol caused by familial hypercholesterolemia and other conditions. The statins resemble mevalonate (Box 21-2) and are competitive inhibitors of HMG-CoA reductase.
An alternative approach to controlling serum cholesterol levels is to activate LXRs, which has the overall effect of decreasing cholesterol absorption and promoting its excretion. This is the mode of action of a drug called ezetimibe. Ezetimibe is not as effective in lowering serum cholesterol as statins. However, in combination with statins, it can lower risk of stroke or heart attack in high-risk patients. Because LXR activation also activates SREBP1C, causing the liver to increase its production of fatty acids and triacylglycerols, new classes of drugs that target only intestinal LXRs are being developed.
Reverse Cholesterol Transport by HDL Counters Plaque Formation and Atherosclerosis
HDL plays a critical role in the reverse cholesterol transport pathway (Fig. 21-47), reducing the potential damage from foam cell buildup. Depleted HDL (low in cholesterol) picks up cholesterol stored in extrahepatic tissues, including foam cells at nascent plaques, and carries it to the liver.
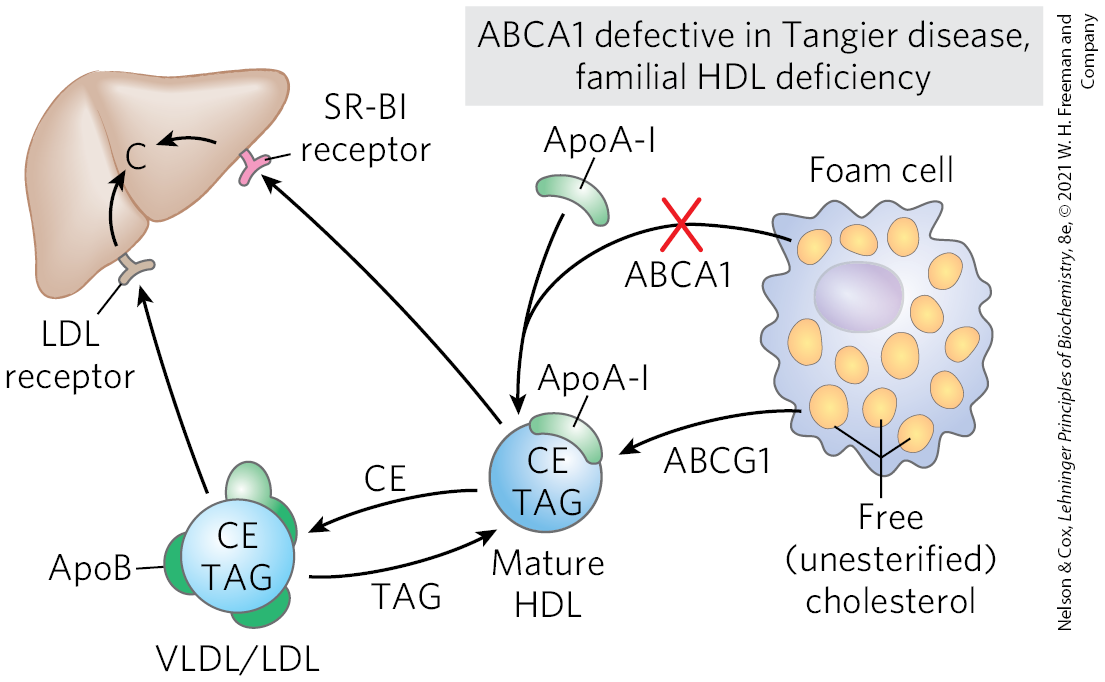
FIGURE 21-47 Reverse cholesterol transport. ApoA-I and HDLs pick up excess cholesterol (C) from peripheral cells, with the participation of ABCA1 and ABCG1 transporters, and return it to the liver. In individuals with genetically defective ABCA1, the failure of reverse cholesterol transport leads to severe and early cardiovascular diseases: Tangier disease and familial HDL deficiency disease. CE, cholesteryl esters; TAG, triacylglycerols. [Information from A. R. Tall et al., Cell Metab. 7:365, 2008, Fig. 1.]
Cholesterol movement out of cells requires transporters. The human genome encodes 48 transporters of the ATP-binding cassette (ABC) class, and about half of these promote lipid transport. Two of them transport cholesterol out of cells. In this process, apoA-I interacts with the transporter ABCA1 in a cholesterol-rich cell. ABCA1 transports a load of cholesterol from inside the cell to the outer surface of the plasma membrane, where lipid-free or lipid-poor apoA-I picks it up, then transports it to the liver. Another transporter, ABCG1, interacts with mature HDL, facilitating the movement of cholesterol out of the cell and into the HDL. This efflux process is particularly critical to reverse cholesterol transport away from foam cells at the sites of plaques in the blood vessels of individuals with cardiovascular disease.
In familial HDL deficiency, HDL levels are very low, and in Tangier disease they are almost undetectable (Fig. 21-47). Both genetic disorders are the result of mutations in the ABCA1 protein. ApoA-I in cholesterol-depleted HDL cannot take up cholesterol from cells that lack ABCA1 protein, and apoA-I and cholesterol-poor HDL are rapidly removed from the blood and destroyed. Both familial HDL deficiency and Tangier disease are very rare (worldwide, fewer than 100 families with Tangier disease are known), but the existence of these diseases establishes a role for ABCA1 and ABCG1 proteins in the regulation of plasma HDL levels.
Steroid Hormones Are Formed by Side-Chain Cleavage and Oxidation of Cholesterol
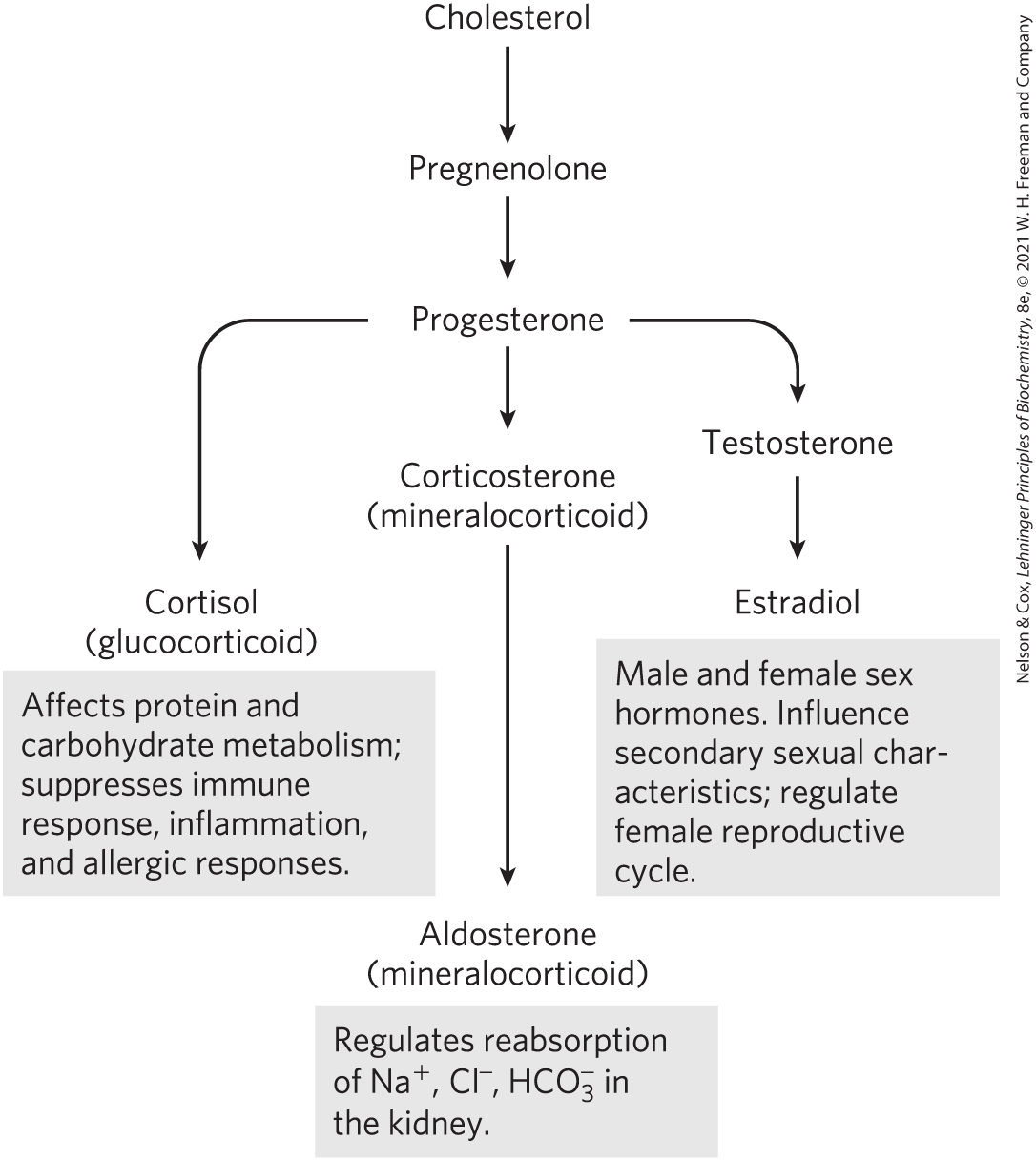
Humans derive all their steroid hormones from cholesterol (Fig. 21-48). Two classes of steroid hormones are synthesized in the cortex of the adrenal gland: mineralocorticoids, which control the reabsorption of inorganic ions (, and ) by the kidney, and glucocorticoids, which help regulate gluconeogenesis and reduce the inflammatory response. Sex hormones are produced in male and female gonads and the placenta. They include progesterone, which regulates the female reproductive cycle, and androgens (such as testosterone) and estrogens (such as estradiol), which influence the development of secondary sexual characteristics in males and females, respectively. Steroid hormones are effective at very low concentrations and are therefore synthesized in relatively small quantities. In comparison with the bile salts, their production consumes relatively little cholesterol.
FIGURE 21-48 Some steroid hormones derived from cholesterol. The structures of some of these compounds are shown in Figure 10-18.
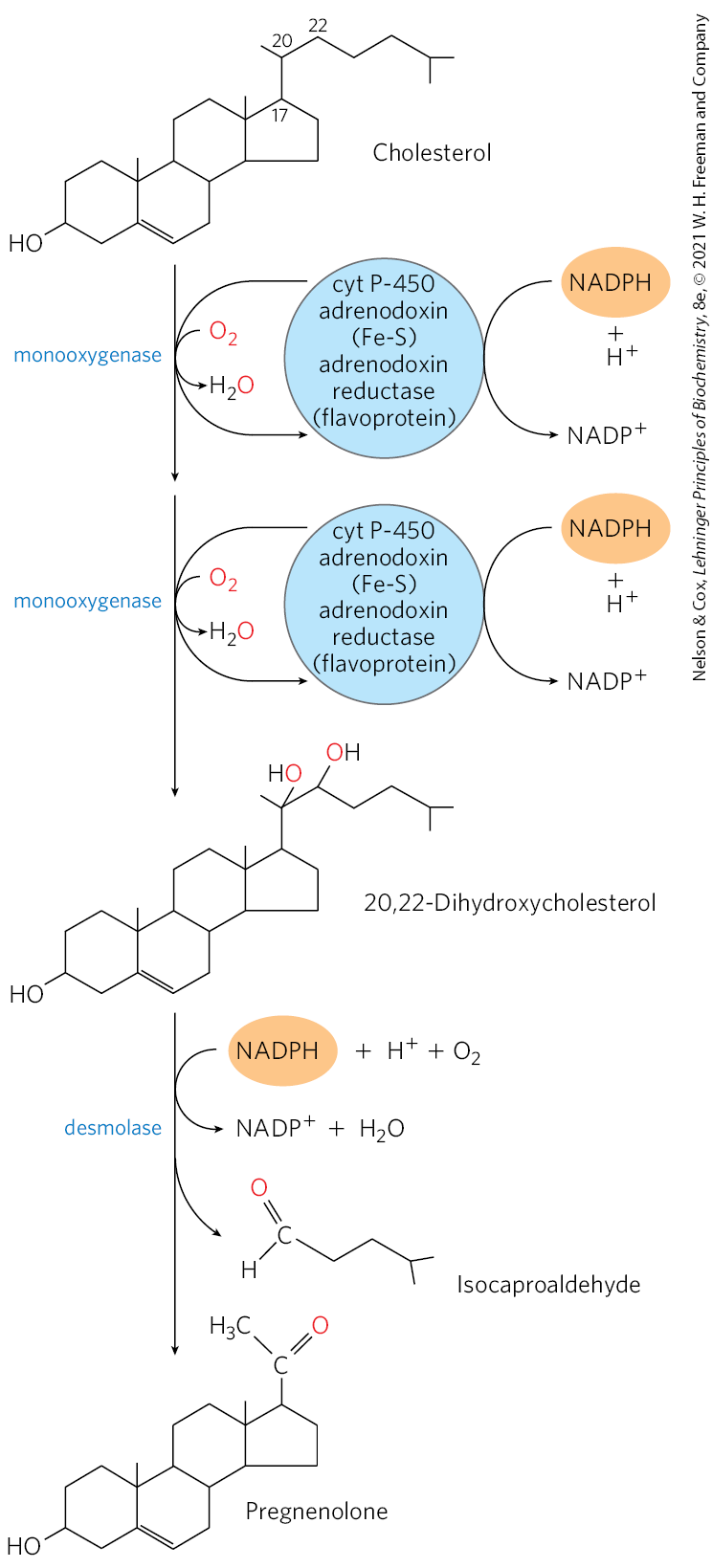
Synthesis of steroid hormones requires removal of some or all of the carbons in the “side chain” on C-17 of the D ring of cholesterol. Side-chain removal takes place in the mitochondria of steroidogenic tissues. Removal requires the hydroxylation of two adjacent carbons in the side chain (C-20 and C-22) followed by cleavage of the bond between them (Fig. 21-49). Formation of the various hormones also requires the introduction of oxygen atoms. All the hydroxylation and oxygenation reactions in steroid biosynthesis are catalyzed by mixed-function oxygenases (see Box 21-1) that use NADPH, , and mitochondrial cytochrome P-450.
FIGURE 21-49 Side-chain cleavage in the synthesis of steroid hormones. Cytochrome P-450 acts as electron carrier in this monooxygenase system that oxidizes adjacent carbons. The process also requires the electron-transferring proteins adrenodoxin and adrenodoxin reductase. This system for cleaving side chains is found in mitochondria of the adrenal cortex, where active steroid production occurs. Pregnenolone is the precursor of all other steroid hormones (see Fig. 21-48).
Intermediates in Cholesterol Biosynthesis Have Many Alternative Fates
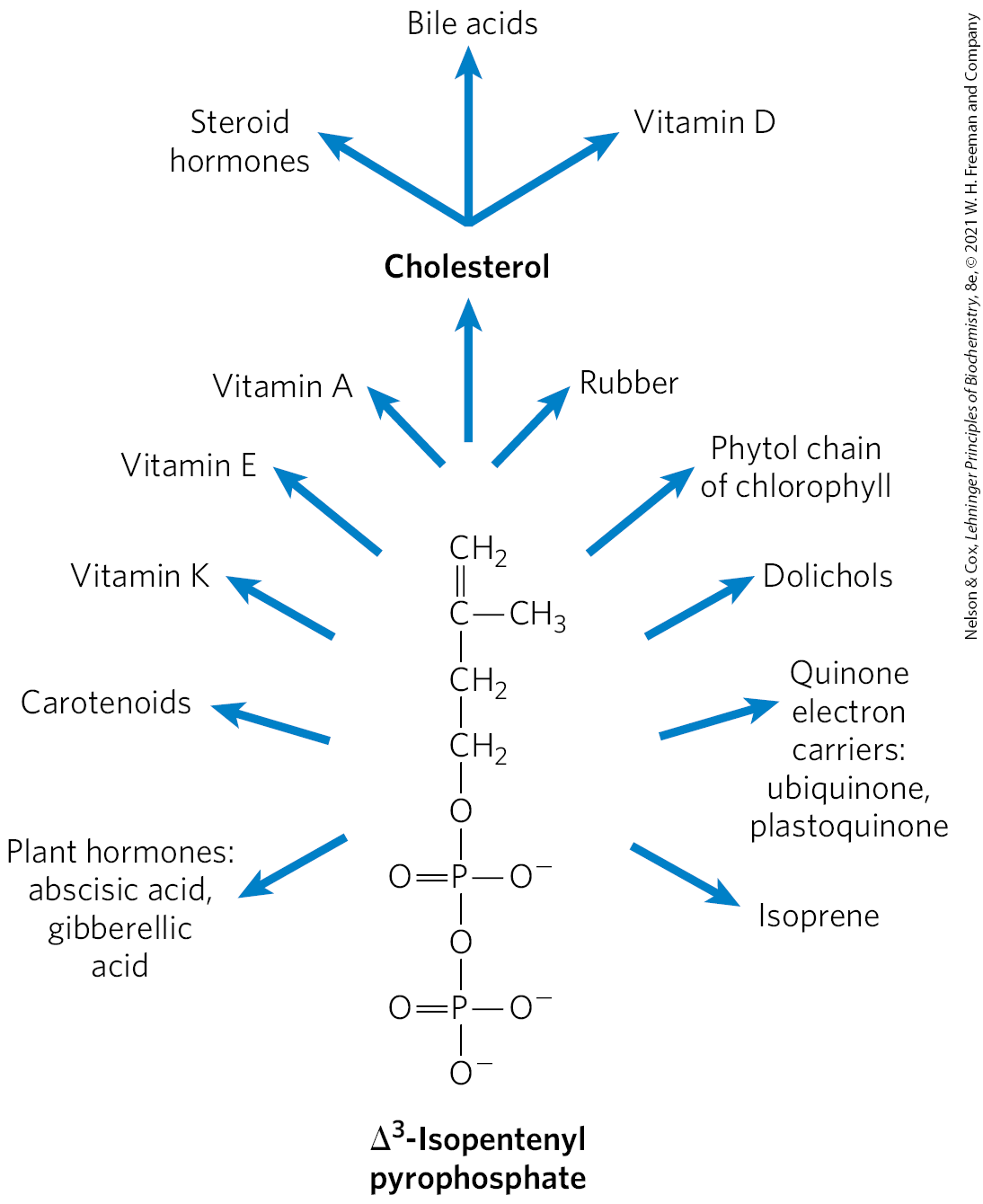
In addition to its role as an intermediate in cholesterol biosynthesis, isopentenyl pyrophosphate is the activated precursor of a huge array of biomolecules with diverse biological roles (Fig. 21-50). They include vitamins A, E, and K; plant pigments such as carotene and the phytol chain of chlorophyll; natural rubber; many essential oils, such as the fragrant principles of lemon oil, eucalyptus, and musk; insect juvenile hormone, which controls metamorphosis; dolichols, which serve as lipid-soluble carriers in complex polysaccharide synthesis; and ubiquinone and plastoquinone, electron carriers in mitochondria and chloroplasts. Collectively, these molecules are called isoprenoids. More than 20,000 different isoprenoid molecules have been discovered in nature, and hundreds of new ones are reported each year.
FIGURE 21-50 Overview of isoprenoid biosynthesis. The structures of most of the end products shown here are given in Chapter 10.
Prenylation, the covalent attachment of an isoprenoid (see Fig. 27-35), is a common mechanism by which proteins are anchored to the inner surface of cellular membranes in mammals (see Fig. 11-16). In some of these proteins, the attached lipid is the 15-carbon farnesyl group; others have the 20-carbon geranylgeranyl group. Different enzymes attach the two types of lipids. It is possible that prenylation targets proteins to different membranes, depending on which lipid is attached. Protein prenylation is another important role for the isoprene derivatives of the pathway to cholesterol.
SUMMARY 21.4 Cholesterol, Steroids, and Isoprenoids: Biosynthesis, Regulation, and Transport
Cholesterol is formed from acetyl-CoA in a complex series of reactions, through the intermediates β-hydroxy-β-methylglutaryl-CoA (HMG-CoA), mevalonate, and two activated isoprenes, dimethylallyl pyrophosphate and isopentenyl pyrophosphate. Condensation of isoprene units produces the noncyclic squalene, which is cyclized to yield the steroid ring system and side chain.
Synthesized primarily in the liver, cholesterol is exported as bile acids, biliary cholesterol, or cholesteryl esters.
Cholesterol and cholesteryl esters are carried in the blood as plasma lipoproteins. VLDL carries cholesterol, cholesteryl esters, and triacylglycerols from the liver to other tissues, where the triacylglycerols are degraded by lipoprotein lipase, converting VLDL to LDL. Cholesterol scavenging and transport back to the liver are mediated by HDL. Much of the lipid carried to the liver is utilized in bile salts.
The LDL, rich in cholesterol and its esters, is taken up by receptor-mediated endocytosis, in which the apolipoprotein B-100 of LDL is recognized by receptors in the plasma membrane.
Cholesterol synthesis and transport are under complex regulation by hormones, cellular cholesterol content, and energy level (AMP concentration). HMG-CoA reductase is regulated allosterically and by covalent modification. Furthermore, a complex of three proteins—Insig, SCAP, and SREBP — sense cholesterol levels and trigger increased synthesis or degradation of HMG-CoA reductase in response. The number of LDL receptors per cell is also regulated by cholesterol content.
Dietary conditions or genetic defects in cholesterol metabolism may lead to atherosclerosis and heart disease.
In reverse cholesterol transport, HDL removes cholesterol from peripheral tissues, carrying it to the liver. By reducing the cholesterol content of foam cells, HDL protects against atherosclerosis.
The steroid hormones (glucocorticoids, mineralocorticoids, and sex hormones) are produced from cholesterol by alteration of the side chain and introduction of oxygen atoms into the steroid ring system. In addition to cholesterol, a wide variety of isoprenoid compounds are derived from mevalonate through condensations of isopentenyl pyrophosphate and dimethylallyl pyrophosphate.
Prenylation of certain proteins targets them for association with cellular membranes and is essential for their biological activity.
 Cholesterol, like long-chain fatty acids, is made from acetyl-CoA. But the assembly plan of cholesterol is quite different from that of long-chain fatty acids. In early experiments, animals were fed acetate labeled with in either the methyl carbon or the carboxyl carbon. The pattern of labeling in the cholesterol isolated from the two groups of animals in these tracer experiments (
Cholesterol, like long-chain fatty acids, is made from acetyl-CoA. But the assembly plan of cholesterol is quite different from that of long-chain fatty acids. In early experiments, animals were fed acetate labeled with in either the methyl carbon or the carboxyl carbon. The pattern of labeling in the cholesterol isolated from the two groups of animals in these tracer experiments (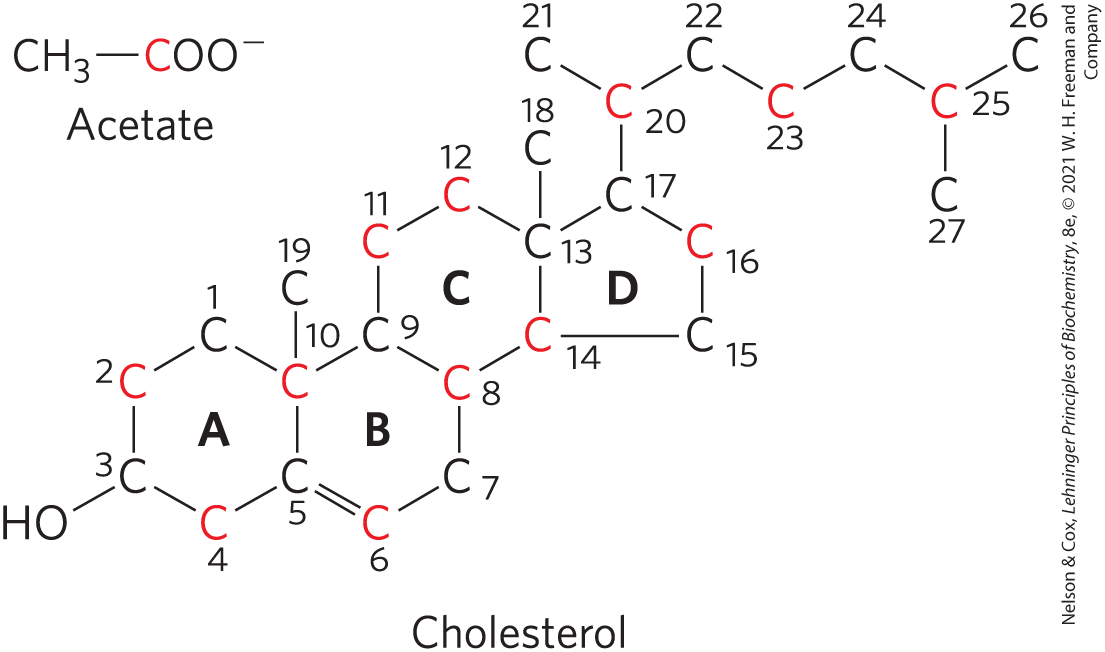
 condensation of three acetate units to form a six-carbon intermediate, mevalonate;
condensation of three acetate units to form a six-carbon intermediate, mevalonate;  conversion of mevalonate to activated isoprene units;
conversion of mevalonate to activated isoprene units;  polymerization of six 5-carbon isoprene units to form the 30-carbon linear squalene; and
polymerization of six 5-carbon isoprene units to form the 30-carbon linear squalene; and  cyclization of squalene to form the four rings of the steroid nucleus, with a further series of changes (oxidations, removal or migration of methyl groups) to produce cholesterol.
cyclization of squalene to form the four rings of the steroid nucleus, with a further series of changes (oxidations, removal or migration of methyl groups) to produce cholesterol.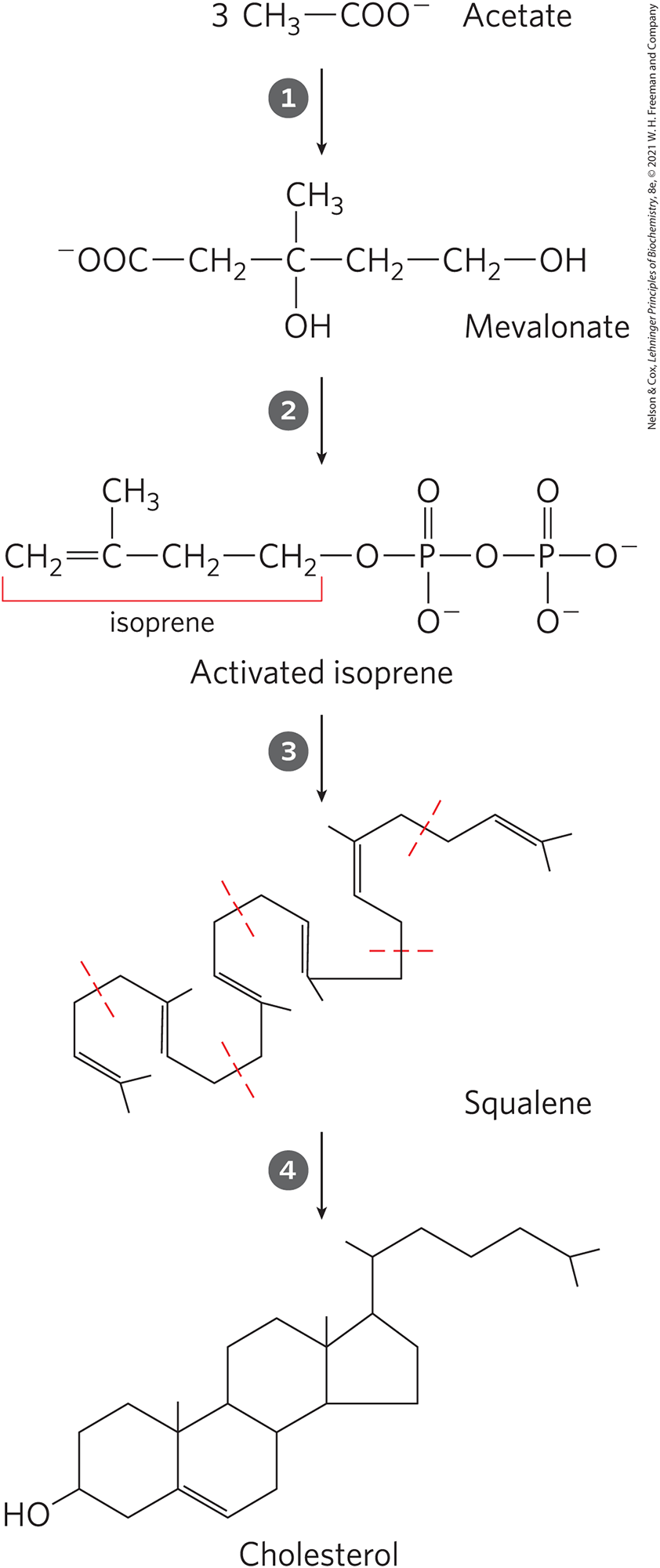
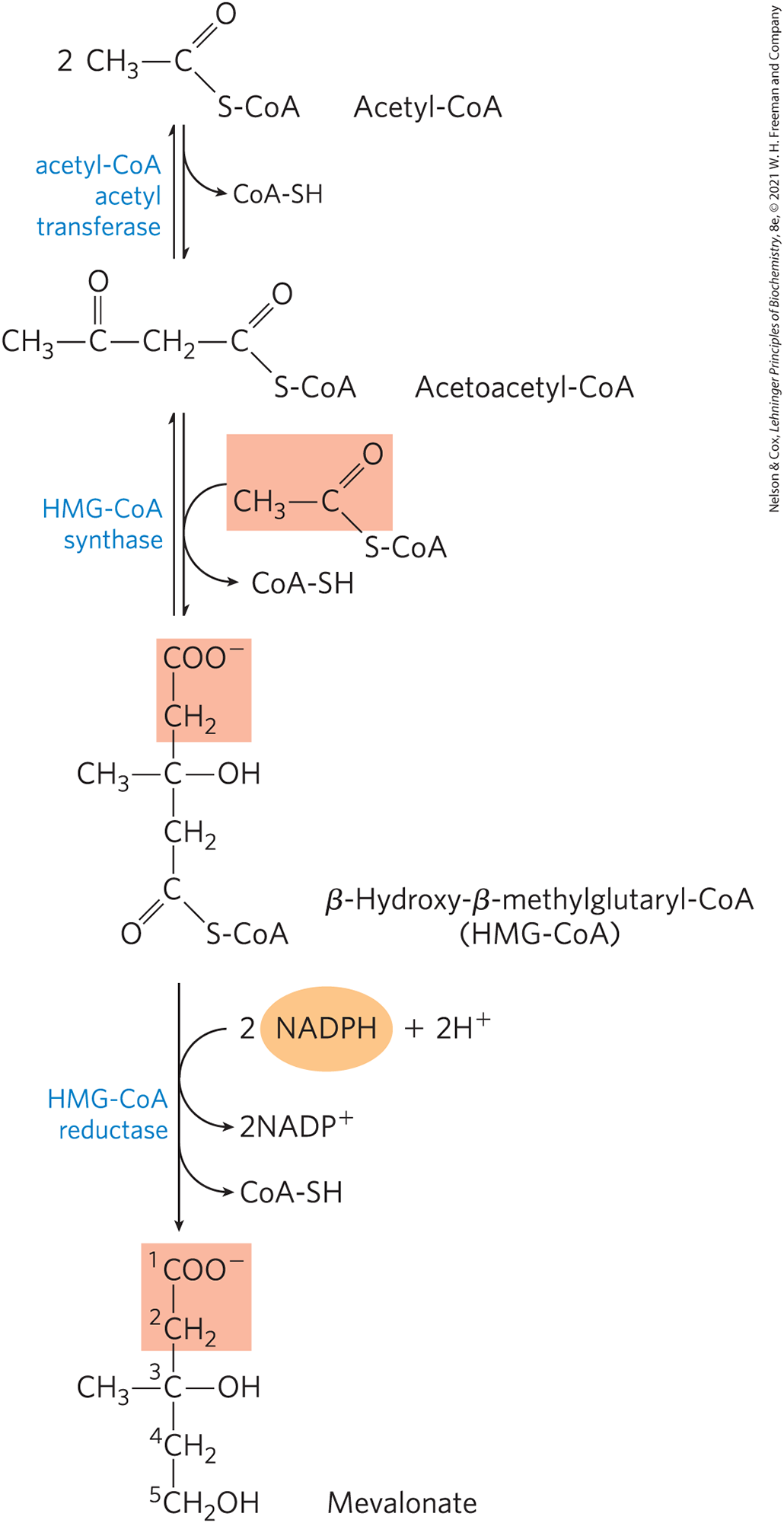
 The third reaction is the committed step: reduction of HMG-CoA to mevalonate, for which two molecules of NADPH each donate two electrons. HMG-CoA reductase, an integral membrane protein of the smooth ER, is the major point of regulation on the pathway to cholesterol, as we shall see.
The third reaction is the committed step: reduction of HMG-CoA to mevalonate, for which two molecules of NADPH each donate two electrons. HMG-CoA reductase, an integral membrane protein of the smooth ER, is the major point of regulation on the pathway to cholesterol, as we shall see.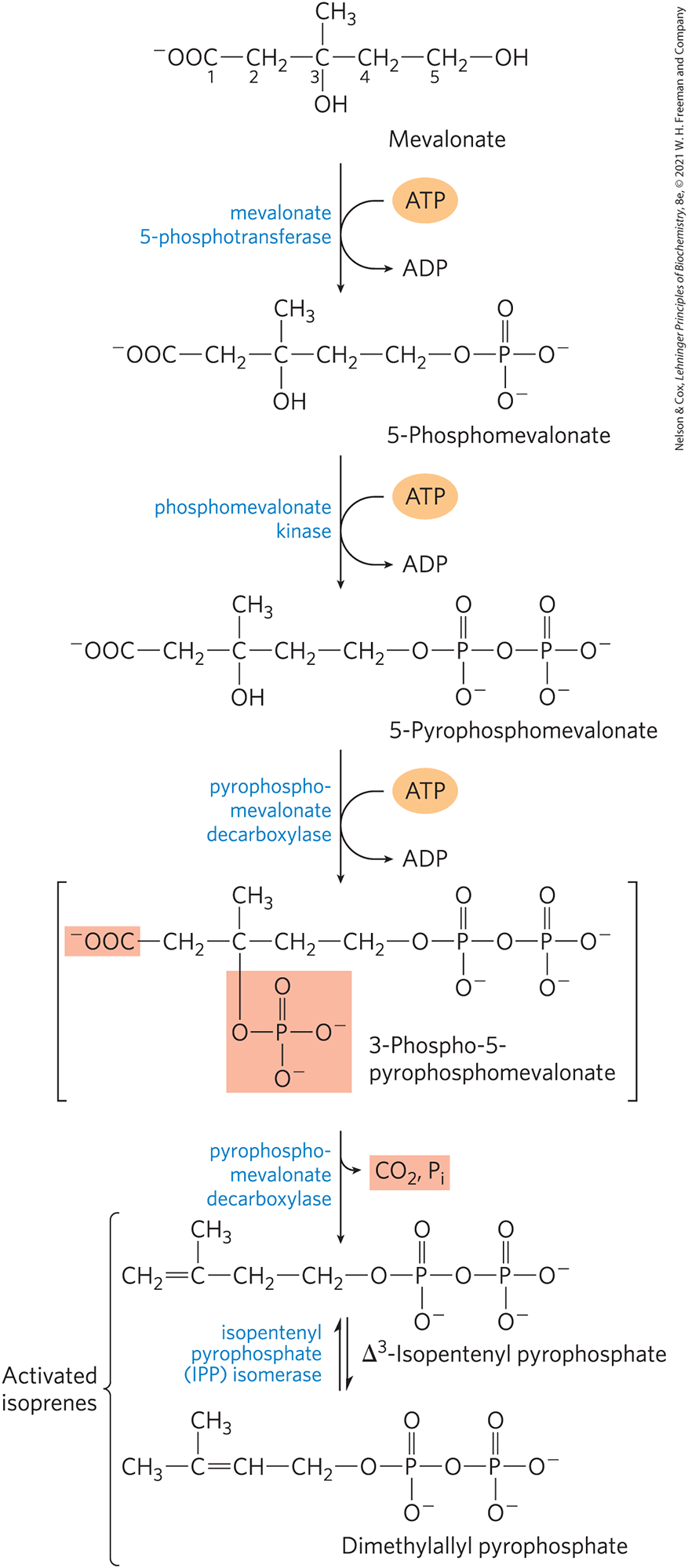

 they are converted to triacylglycerols or cholesteryl esters in the liver and packaged with specific apolipoproteins into VLDL. Excess carbohydrate in the diet can also be converted to triacylglycerols in the liver and exported as VLDL. In addition to triacylglycerols and cholesteryl esters, VLDL contains apoB-100, apoC-I, apoC-II, apoC-III, and apoE (
they are converted to triacylglycerols or cholesteryl esters in the liver and packaged with specific apolipoproteins into VLDL. Excess carbohydrate in the diet can also be converted to triacylglycerols in the liver and exported as VLDL. In addition to triacylglycerols and cholesteryl esters, VLDL contains apoB-100, apoC-I, apoC-II, apoC-III, and apoE ( In the capillaries of these tissues, apoC-II activates lipoprotein lipase, which catalyzes the release of free fatty acids from triacylglycerols in the VLDL. Adipocytes take up these fatty acids, reconvert them to triacylglycerols, and store the products in intracellular lipid droplets; myocytes, in contrast, primarily oxidize the fatty acids to supply energy. When the insulin level is high (after a meal), VLDL serves primarily to convey lipids from the diet to adipose tissue for storage. In the fasting state between meals, the fatty acids used to produce VLDL in the liver originate primarily from adipose tissue, and the principal VLDL target is myocytes of the heart and skeletal muscle.
In the capillaries of these tissues, apoC-II activates lipoprotein lipase, which catalyzes the release of free fatty acids from triacylglycerols in the VLDL. Adipocytes take up these fatty acids, reconvert them to triacylglycerols, and store the products in intracellular lipid droplets; myocytes, in contrast, primarily oxidize the fatty acids to supply energy. When the insulin level is high (after a meal), VLDL serves primarily to convey lipids from the diet to adipose tissue for storage. In the fasting state between meals, the fatty acids used to produce VLDL in the liver originate primarily from adipose tissue, and the principal VLDL target is myocytes of the heart and skeletal muscle. LDL carries cholesterol to extrahepatic tissues such as muscle, adrenal glands, and adipose tissue. These tissues have plasma membrane LDL receptors that recognize apoB-100 and mediate uptake of cholesterol and cholesteryl esters.
LDL carries cholesterol to extrahepatic tissues such as muscle, adrenal glands, and adipose tissue. These tissues have plasma membrane LDL receptors that recognize apoB-100 and mediate uptake of cholesterol and cholesteryl esters.  LDL also delivers cholesterol to macrophages, sometimes converting them into foam cells (see
LDL also delivers cholesterol to macrophages, sometimes converting them into foam cells (see  LDL not taken up by peripheral tissues and cells returns to the liver and is taken up via LDL receptors in the hepatocyte plasma membrane. Cholesterol that enters hepatocytes by this path may be incorporated into membranes, converted to bile acids, or reesterified by ACAT (
LDL not taken up by peripheral tissues and cells returns to the liver and is taken up via LDL receptors in the hepatocyte plasma membrane. Cholesterol that enters hepatocytes by this path may be incorporated into membranes, converted to bile acids, or reesterified by ACAT ( originates
originates Nascent
Nascent Mature
Mature Some of the cholesteryl esters in HDL can also be transferred to LDL by the cholesteryl ester transfer protein. The HDL circuit is reverse cholesterol transport (purple arrows in
Some of the cholesteryl esters in HDL can also be transferred to LDL by the cholesteryl ester transfer protein. The HDL circuit is reverse cholesterol transport (purple arrows in  Bile salts are reabsorbed by the liver and recirculate through the gallbladder in this enterohepatic circulation (green arrows in
Bile salts are reabsorbed by the liver and recirculate through the gallbladder in this enterohepatic circulation (green arrows in 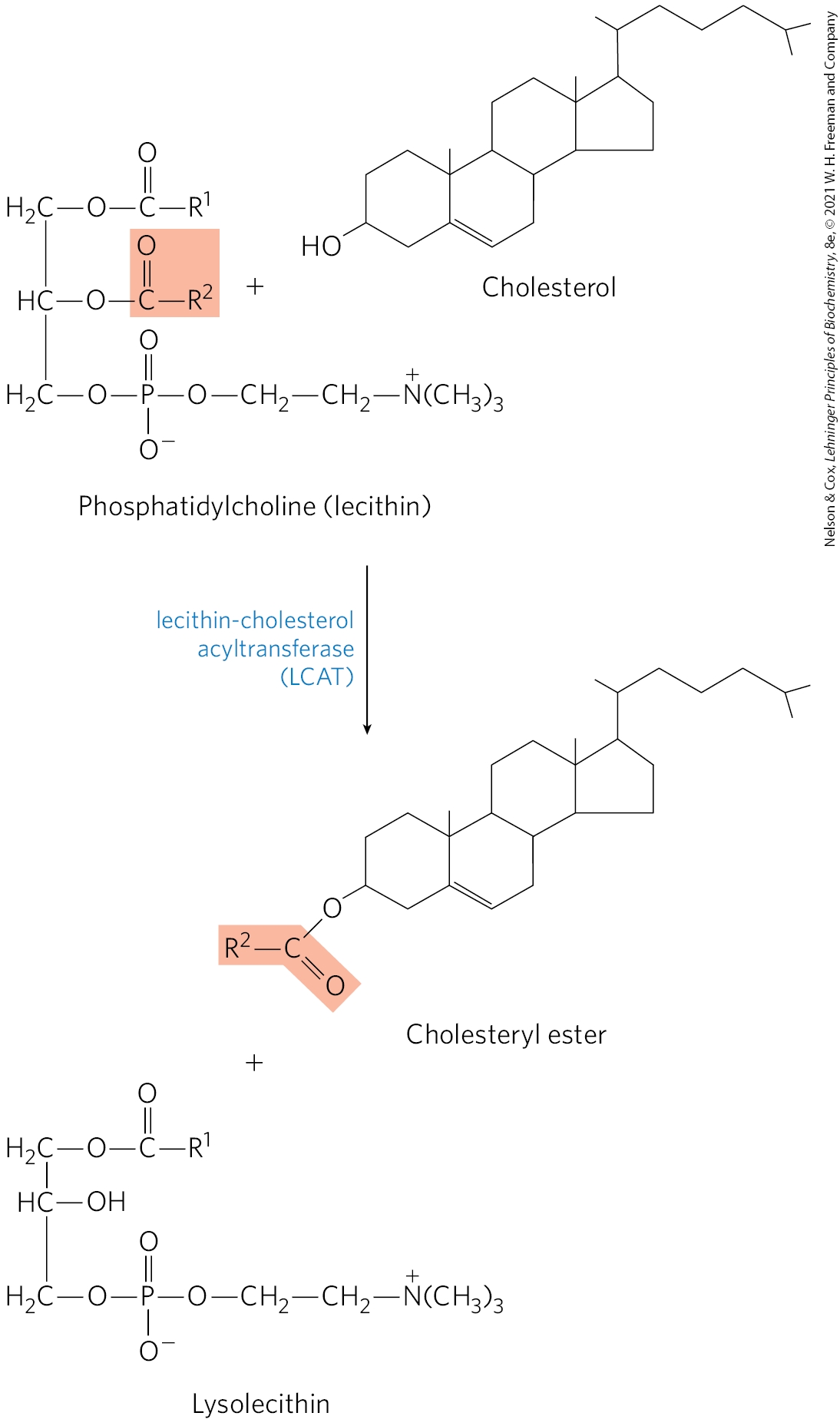
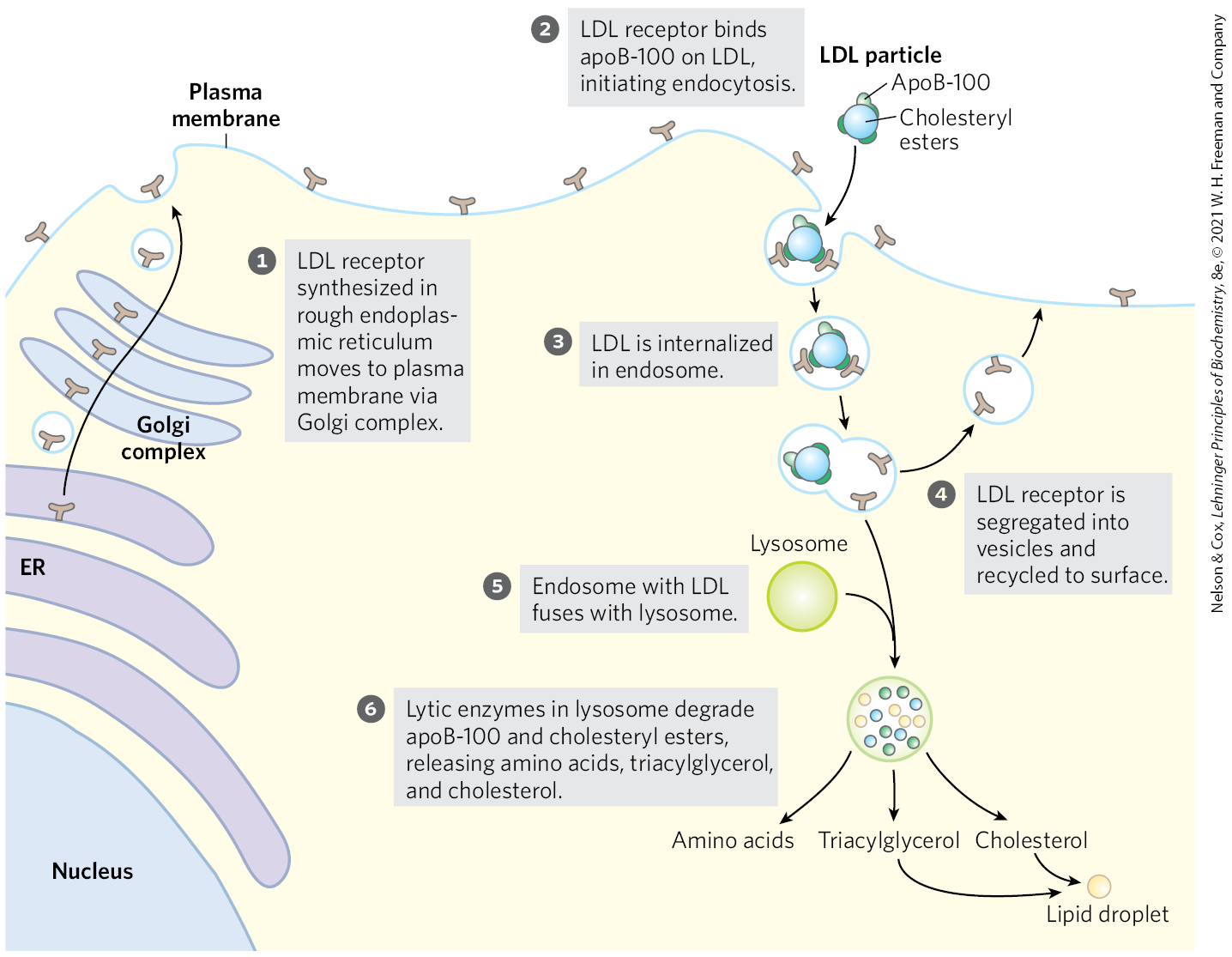
 This pathway for the transport of cholesterol in blood and its receptor-mediated endocytosis by target tissues was elucidated by Michael Brown and Joseph Goldstein. They discovered that individuals with the genetic disease familial hypercholesterolemia (FH) have mutations in the LDL receptor that prevent the normal uptake of LDL by liver and peripheral tissues. The result of defective LDL uptake is very high blood levels of LDL (and of the cholesterol it carries). Individuals with FH have a greatly increased probability of developing atherosclerosis, a disease of the cardiovascular system in which blood vessels are occluded by cholesterol-rich plaques (see
This pathway for the transport of cholesterol in blood and its receptor-mediated endocytosis by target tissues was elucidated by Michael Brown and Joseph Goldstein. They discovered that individuals with the genetic disease familial hypercholesterolemia (FH) have mutations in the LDL receptor that prevent the normal uptake of LDL by liver and peripheral tissues. The result of defective LDL uptake is very high blood levels of LDL (and of the cholesterol it carries). Individuals with FH have a greatly increased probability of developing atherosclerosis, a disease of the cardiovascular system in which blood vessels are occluded by cholesterol-rich plaques (see 

 Cholesterol is formed from acetyl-CoA in a complex series of reactions, through the intermediates β-hydroxy-β-methylglutaryl-CoA (HMG-CoA), mevalonate, and two activated isoprenes, dimethylallyl pyrophosphate and isopentenyl pyrophosphate. Condensation of isoprene units produces the noncyclic squalene, which is cyclized to yield the steroid ring system and side chain.
Cholesterol is formed from acetyl-CoA in a complex series of reactions, through the intermediates β-hydroxy-β-methylglutaryl-CoA (HMG-CoA), mevalonate, and two activated isoprenes, dimethylallyl pyrophosphate and isopentenyl pyrophosphate. Condensation of isoprene units produces the noncyclic squalene, which is cyclized to yield the steroid ring system and side chain.