13.2 Chemical Logic and Common Biochemical Reactions
The biological energy transductions we are concerned with in this book are chemical reactions. Cellular chemistry does not encompass every kind of reaction learned in a typical organic chemistry course. Which reactions take place in biological systems and which do not is determined by (1) their relevance to that particular metabolic system and (2) their rates. Both considerations play major roles in shaping the metabolic pathways we consider throughout the rest of the book. A relevant reaction is one that makes use of an available substrate and converts it to a useful product. However, even a potentially relevant reaction may not occur. Some chemical transformations are too slow (have activation energies that are too high) to contribute to living systems, even with the aid of powerful enzyme catalysts. The reactions that do occur in cells represent a toolbox that evolution has used to construct metabolic pathways that circumvent the “impossible” reactions. Learning to recognize the plausible reactions can be a great aid in developing a command of biochemistry.
Even so, the number of metabolic transformations taking place in a typical cell can seem overwhelming. Most cells have the capacity to carry out thousands of specific, enzyme-catalyzed reactions: for example, transformation of a simple nutrient such as glucose into amino acids, nucleotides, or lipids; extraction of energy from fuels by oxidation; and polymerization of monomeric subunits into macromolecules.
Biochemical Reactions Occur in Repeating Patterns
To study these reactions, some organization is essential. There are patterns within the chemistry of life; you do not need to learn every individual reaction to comprehend the molecular logic of biochemistry. Most of the reactions in living cells fall into one of five general categories: (1) reactions that make or break carbon–carbon bonds; (2) internal rearrangements, isomerizations, and eliminations; (3) free-radical reactions; (4) group transfers; and (5) oxidation-reductions. We discuss each of these in more detail below and refer to some examples of each type in later chapters. Note that the five reaction types are not mutually exclusive; for example, an isomerization reaction may involve a free-radical intermediate.
Before proceeding, however, we should review two basic chemical principles. First, a covalent bond consists of a shared pair of electrons, and the bond can be broken in two general ways (Fig. 13-1). In homolytic cleavage, each atom leaves the bond as a radical, carrying one unpaired electron. In heterolytic cleavage, which is more common, one atom retains both bonding electrons. The species most often generated when and bonds are cleaved are illustrated in Figure 13-1. Carbanions, carbocations, and hydride ions are highly unstable; this instability shapes the chemistry of these ions, as we shall see.
FIGURE 13-1 Two mechanisms for cleavage of a or bond. In a homolytic cleavage, each atom keeps one of the bonding electrons, resulting in the formation of carbon radicals (carbons having unpaired electrons) or uncharged hydrogen atoms. In a heterolytic cleavage, one of the atoms retains both bonding electrons. This can result in the formation of carbanions, carbocations, protons, or hydride ions.
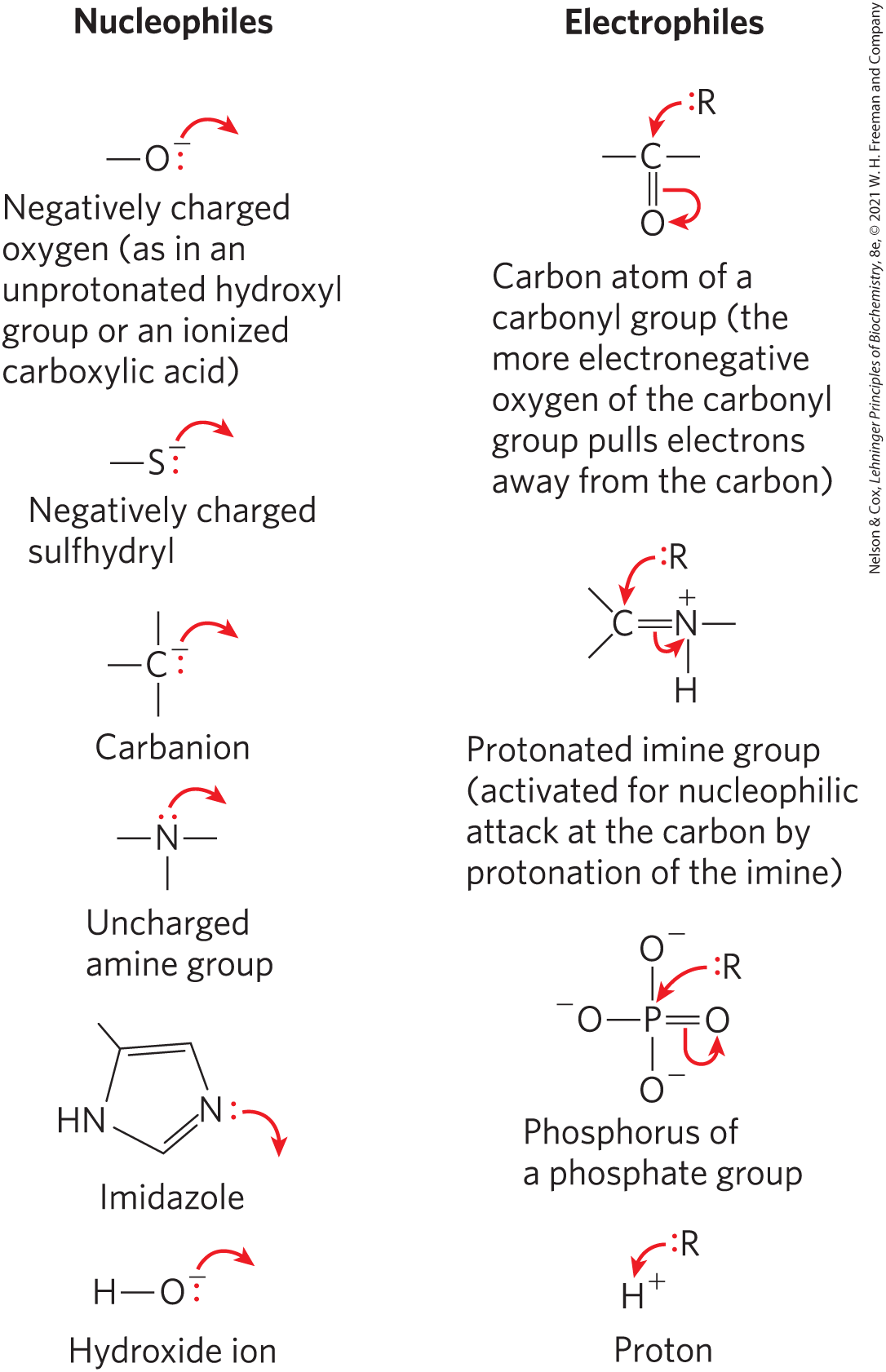
The second basic principle is that many biochemical reactions involve interactions between nucleophiles (functional groups rich in and capable of donating electrons) and electrophiles (electron-deficient functional groups that seek electrons). Nucleophiles combine with and give up electrons to electrophiles. Common biological nucleophiles and electrophiles are shown in Figure 13-2. Note that a carbon atom can act as either a nucleophile or an electrophile, depending on which bonds and functional groups surround it.
FIGURE 13-2 Common nucleophiles and electrophiles in biochemical reactions. Chemical reaction mechanisms, which trace the formation and breakage of covalent bonds, are communicated with dots and curved arrows, a convention known informally as “electron pushing.” A covalent bond consists of a shared pair of electrons. Nonbonded electrons important to the reaction mechanism are designated by dots (:). Curved arrows () represent the movement of electron pairs. For movement of a single electron (as in a free-radical reaction), a single-headed (fishhook-type) arrow is used (). Most reaction steps involve an unshared electron pair.
Reactions That Make or Break Carbon–Carbon Bonds
Heterolytic cleavage of a bond yields a carbanion and a carbocation (Fig. 13-1). Conversely, the formation of a bond involves the combination of a nucleophilic carbanion and an electrophilic carbocation. Carbanions and carbocations are generally so unstable that their formation as reaction intermediates can be energetically unfeasible, even with enzyme catalysts. For the purpose of cellular biochemistry, they are impossible reactions — unless chemical assistance is provided in the form of functional groups containing electronegative atoms (O and N) that can alter the electronic structure of adjacent carbon atoms so as to stabilize and facilitate the formation of carbanion and carbocation intermediates.
Carbonyl groups are particularly important in the chemical transformations of metabolic pathways. The carbon of a carbonyl group has a partial positive charge due to the electron-withdrawing property of the carbonyl oxygen, and so is an electrophilic carbon (Fig. 13-3a). A carbonyl group can thus facilitate the formation of a carbanion on an adjoining carbon by delocalizing the carbanion’s negative charge (Fig. 13-3b). An imine group (see Fig. 1-14) can serve a similar function (Fig. 13-3c). The capacity of carbonyl and imine groups to delocalize electrons can be further enhanced by a general acid catalyst or by a metal ion such as (Fig. 13-3d).
FIGURE 13-3 Chemical properties of carbonyl groups. (a) The carbon atom of a carbonyl group is an electrophile by virtue of the electron-withdrawing capacity of the electronegative oxygen atom, which results in a structure in which the carbon has a partial positive charge. (b) Within a molecule, delocalization of electrons into a carbonyl group stabilizes a carbanion on an adjacent carbon, facilitating its formation. (c) Imines function much like carbonyl groups in facilitating electron withdrawal. (d) Carbonyl groups do not always function alone; their capacity as electron sinks often is augmented by interaction with either a metal ion (, such as ) or a general acid (HA).
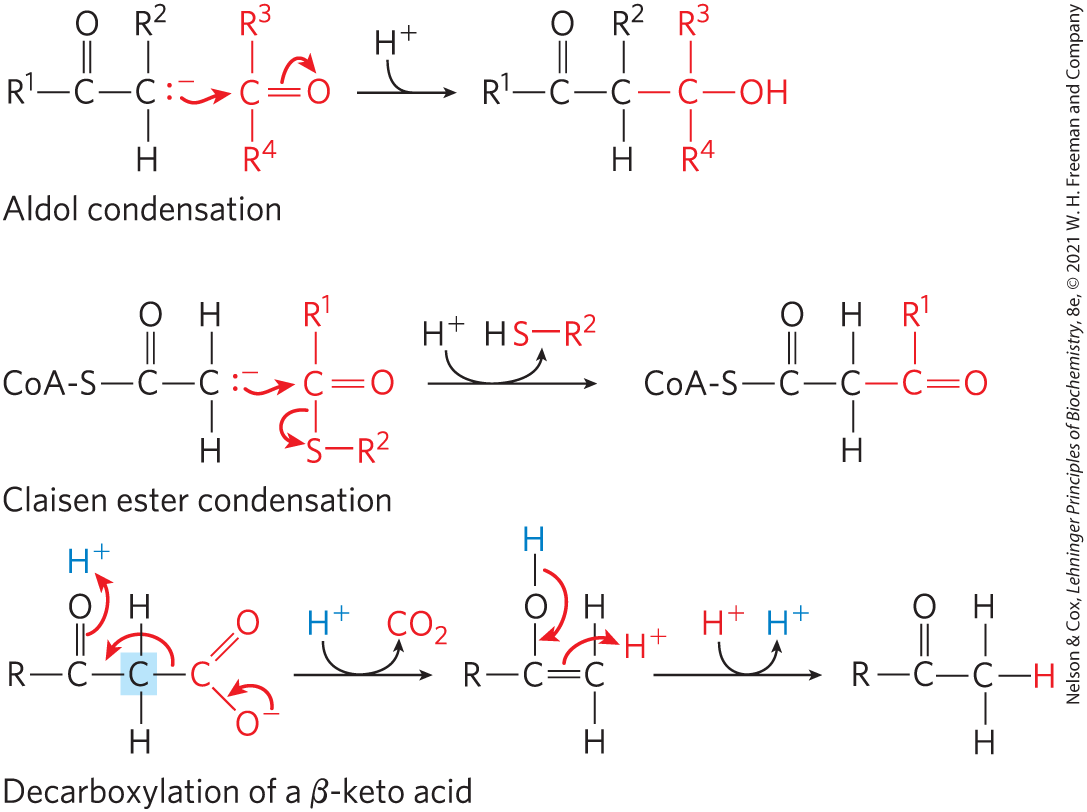
The importance of a carbonyl group is evident in three major classes of reactions in which bonds are formed or broken (Fig. 13-4): aldol condensations, Claisen ester condensations, and decarboxylations. In each type of reaction, a carbanion intermediate is stabilized by a carbonyl group, and in many cases another carbonyl provides the electrophile with which the nucleophilic carbanion reacts.
FIGURE 13-4 Some common reactions that form and break bonds in biological systems. For both the aldol condensation and the Claisen condensation, a carbanion serves as nucleophile and the carbon of a carbonyl group serves as electrophile. The carbanion is stabilized in each case by another carbonyl at the adjoining carbon. In the decarboxylation reaction, a carbanion is formed on the carbon shaded blue as the leaves. The reaction would not occur at an appreciable rate without the stabilizing effect of the carbonyl adjacent to the carbanion carbon. Wherever a carbanion is shown, a stabilizing resonance with the adjacent carbonyl, as shown in Figure 13-3b, is assumed. An imine (Fig. 13-3c) or other electron-withdrawing group (including certain enzymatic cofactors such as pyridoxal) can replace the carbonyl group in the stabilization of carbanions.
An aldol condensation is a common route to the formation of a bond; the aldolase reaction, which converts a six-carbon compound to two three-carbon compounds in glycolysis, is an aldol condensation in reverse (see Fig. 14-5). In a Claisen condensation, the carbanion is stabilized by the carbonyl of an adjacent thioester; an example is the synthesis of citrate in the citric acid cycle (see Fig. 16-9). Decarboxylation also commonly involves the formation of a carbanion stabilized by a carbonyl group; the acetoacetate decarboxylase reaction that occurs in the formation of ketone bodies during fatty acid catabolism provides an example (see Fig. 17-16). Entire metabolic pathways are organized around the introduction of a carbonyl group in a particular location so that a nearby carbon–carbon bond can be formed or cleaved. In some reactions, an imine or a specialized cofactor such as pyridoxal phosphate plays the electron-withdrawing role, instead of a carbonyl group.
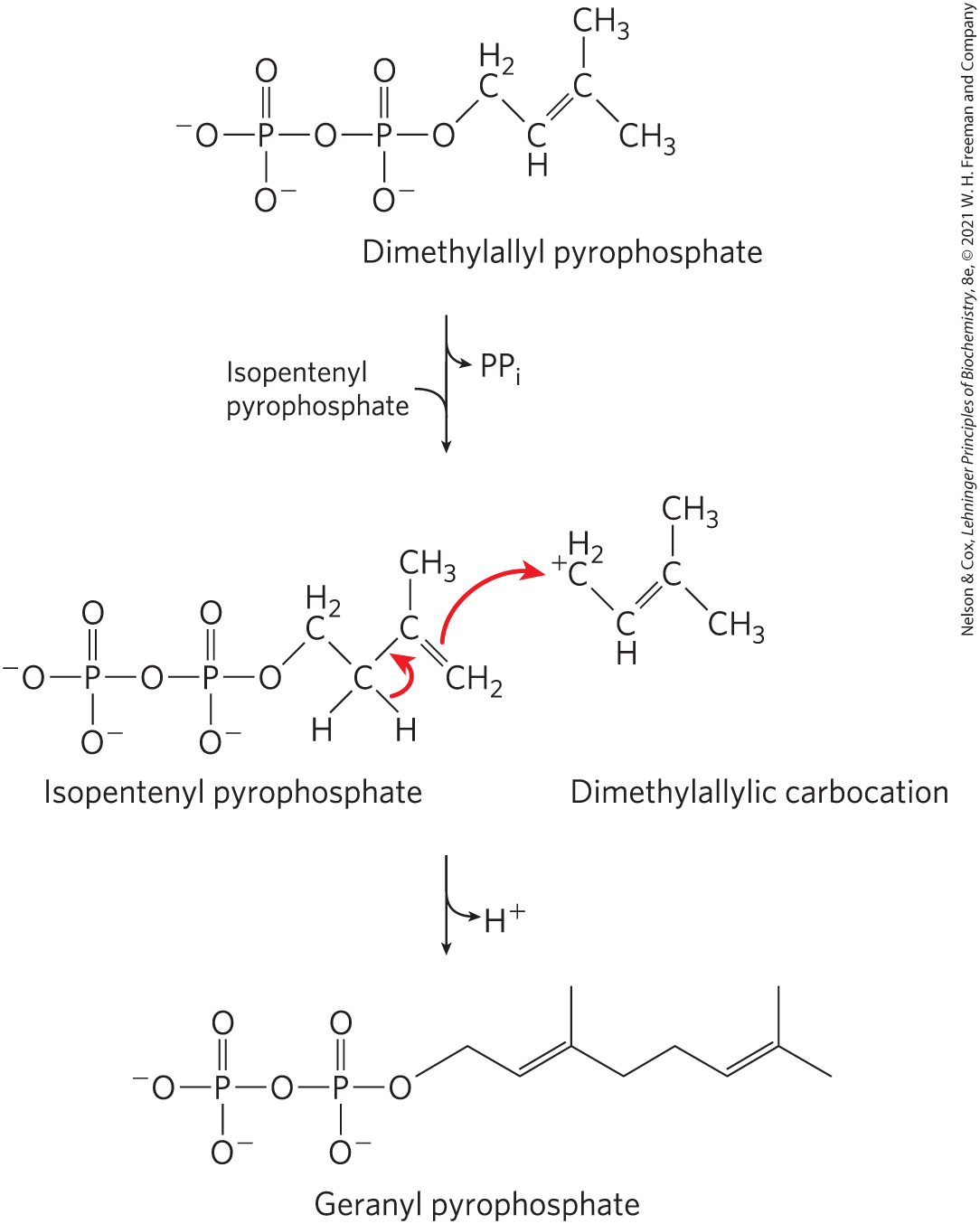
The carbocation intermediate occurring in some reactions that form or cleave bonds is generated by the elimination of an excellent leaving group, such as pyrophosphate (see “Group Transfer Reactions” below). An example is the prenyltransferase reaction (Fig. 13-5), an early step in the pathway of cholesterol biosynthesis.
FIGURE 13-5 Carbocations in carbon–carbon bond formation. In one of the early steps in cholesterol biosynthesis, the enzyme prenyltransferase catalyzes condensation of isopentenyl pyrophosphate and dimethylallyl pyrophosphate to form geranyl pyrophosphate (see Fig. 21-36). The reaction is initiated by elimination of pyrophosphate from the dimethylallyl pyrophosphate to generate a carbocation, stabilized by resonance with the adjacent bond.
Internal Rearrangements, Isomerizations, and Eliminations
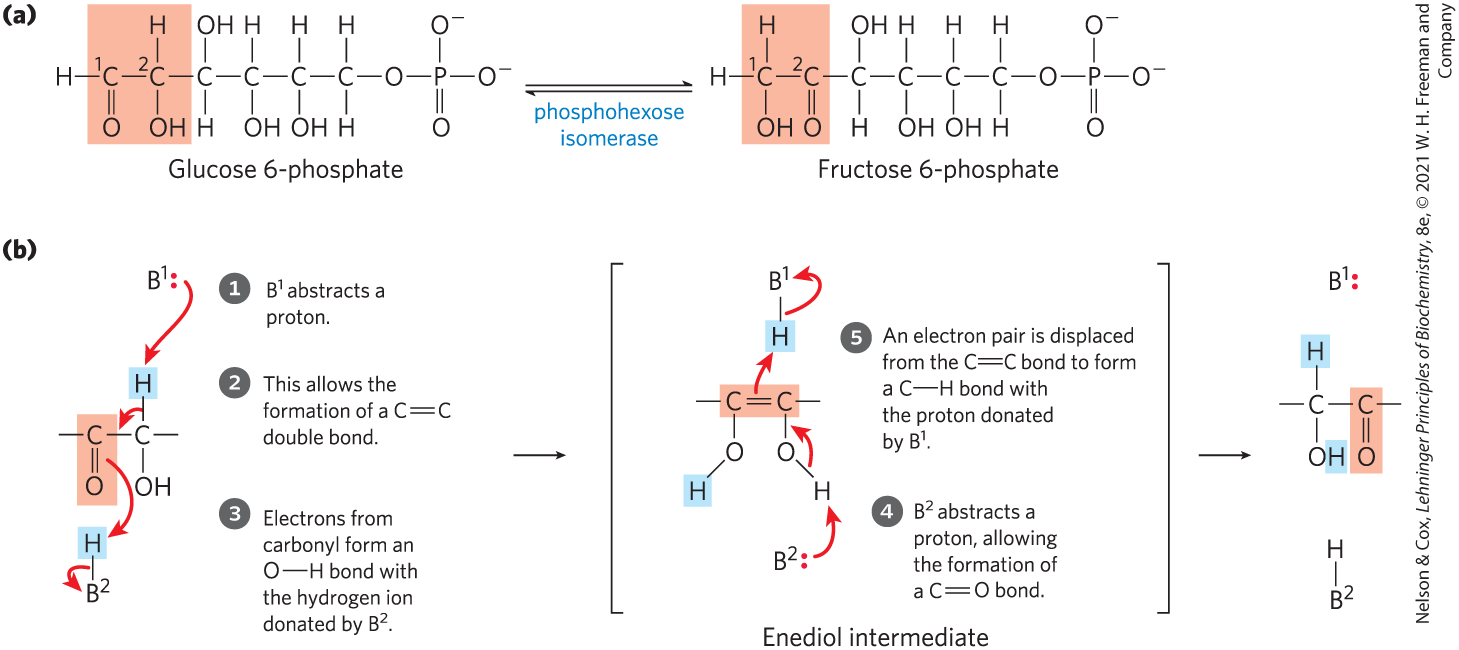
Another common type of cellular reaction is an intramolecular rearrangement in which redistribution of electrons results in alterations of many different types without a change in the overall oxidation state of the molecule. For example, different groups in a molecule may undergo oxidation-reduction, with no net change in oxidation state of the molecule; groups at a double bond may undergo a cis-trans rearrangement; or the positions of double bonds may be transposed. An example of an isomerization entailing internal oxidation-reduction is the formation of fructose 6-phosphate from glucose 6-phosphate in glycolysis (Fig. 13-6; this reaction is discussed in detail in Chapter 14): C-1 is reduced (aldehyde to alcohol) and C-2 is oxidized (alcohol to ketone). Figure 13-6b shows the details of the electron movements in this type of isomerization. A cis-trans rearrangement is illustrated by the prolyl cis-trans isomerase reaction in the folding of certain proteins (see p. 133). A simple transposition of a bond occurs during metabolism of oleic acid, a common fatty acid (see Fig. 17-10). Some spectacular examples of double-bond repositioning occur in the biosynthesis of cholesterol (see Fig. 21-37).
FIGURE 13-6 Isomerization and elimination reactions. (a) The conversion of glucose 6-phosphate to fructose 6-phosphate, a reaction of sugar metabolism catalyzed by phosphohexose isomerase. (b) This reaction proceeds through an enediol intermediate. Light red screens follow the path of oxidation from left to right. and are ionizable groups on the enzyme; they are capable of donating and accepting protons (acting as general acids or general bases) as the reaction proceeds.
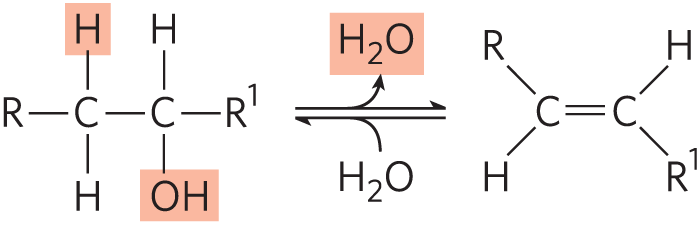
An example of an elimination reaction that does not affect overall oxidation state is the loss of water from an alcohol, resulting in the introduction of a bond:
Similar reactions can result from eliminations in amines.
Free-Radical Reactions
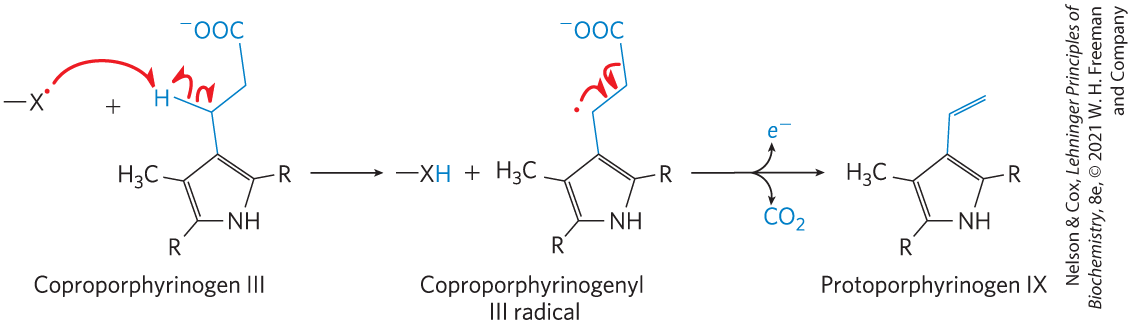
Once thought to be rare, the homolytic cleavage of covalent bonds to generate free radicals has now been found in a wide range of biochemical processes. These include isomerizations that make use of adenosylcobalamin or S-adenosylmethionine, which are initiated with a -deoxyadenosyl radical (see the methylmalonyl-CoA mutase reaction in Box 17-2); certain radical-initiated decarboxylation reactions (Fig. 13-7); some reductase reactions, such as that catalyzed by ribonucleotide reductase (see Fig. 22-43); and some rearrangement reactions, such as that catalyzed by DNA photolyase (see Fig. 25-25).
FIGURE 13-7 A free radical–initiated decarboxylation reaction. The biosynthesis of heme in Escherichia coli includes a decarboxylation step in which propionyl side chains on the coproporphyrinogen III intermediate are converted to the vinyl side chains of protoporphyrinogen IX. When the bacteria are grown anaerobically the enzyme oxygen-independent coproporphyrinogen III oxidase, also called HemN protein, promotes decarboxylation via the free-radical mechanism shown here. The acceptor of the released electron is not known. For simplicity, only the relevant portions of the large coproporphyrinogen III and protoporphyrinogen molecules are shown; the entire structures are given in Figure 22-26. When E. coli is grown in the presence of oxygen, this reaction is an oxidative decarboxylation and is catalyzed by a different enzyme. [Information from G. Layer et al., Curr. Opin. Chem. Biol. 8:468, 2004, Fig. 4.]
Group Transfer Reactions
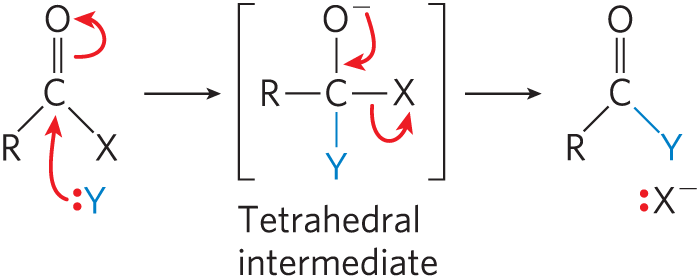
The transfer of acyl, glycosyl, and phosphoryl groups from one nucleophile to another is common in living cells. Acyl group transfer generally involves the addition of a nucleophile to the carbonyl carbon of an acyl group to form a tetrahedral intermediate:
The chymotrypsin reaction is one example of acyl group transfer (see Fig. 6-27). Glycosyl group transfers involve nucleophilic substitution at C-1 of a sugar ring, which is the central atom of an acetal. In principle, the substitution could proceed by an or pathway.
Phosphoryl group transfers play a special role in metabolic pathways, and these transfer reactions are discussed in detail in Section 13.3. A general theme in metabolism is the attachment of a good leaving group to a metabolic intermediate to “activate” the intermediate for subsequent reaction. Among the better leaving groups in nucleophilic substitution reactions are inorganic orthophosphate (the ionized form of at neutral pH, a mixture of and , commonly abbreviated ) and inorganic pyrophosphate , abbreviated ; esters and anhydrides of phosphoric acid are effectively activated for reaction. Nucleophilic substitution is made more favorable by the attachment of a phosphoryl group to an otherwise poor leaving group such as . Nucleophilic substitutions in which the phosphoryl group serves as a leaving group occur in hundreds of metabolic reactions.
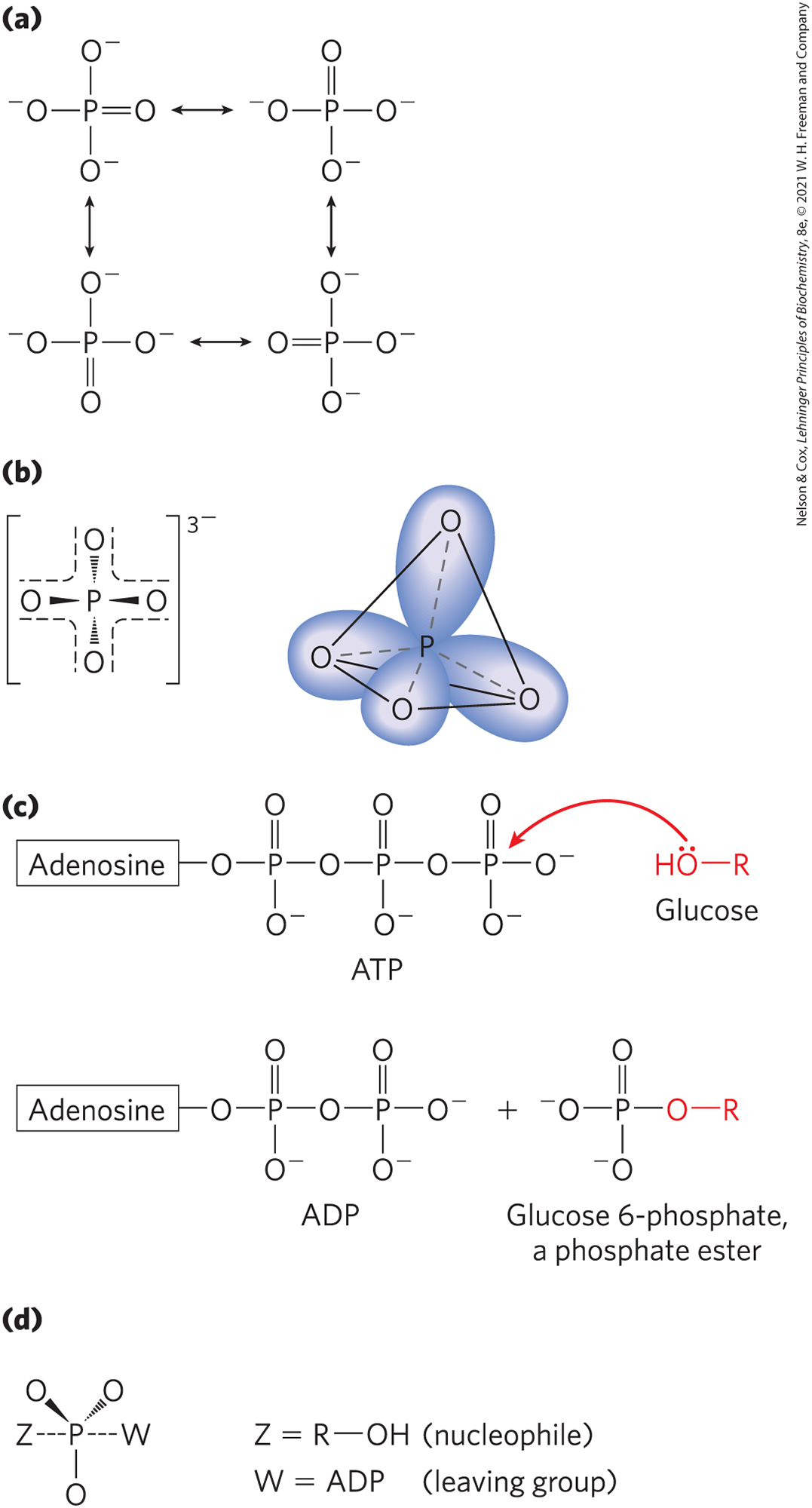
Phosphorus can form five covalent bonds. The conventional representation of (Fig. 13-8a), with three bonds and one bond, is a convenient but inaccurate picture. In , four equivalent phosphorus–oxygen bonds share some double-bond character, and the anion has a tetrahedral structure (Fig. 13-8b). Because oxygen is more electronegative than phosphorus, the sharing of electrons is unequal: the central phosphorus bears a partial positive charge and can therefore act as an electrophile. In a great many metabolic reactions, a phosphoryl group is transferred from ATP to an alcohol, forming a phosphate ester (Fig. 13-8c), or to a carboxylic acid, forming a mixed anhydride. When a nucleophile attacks the electrophilic phosphorus atom in ATP, a relatively stable pentacovalent structure forms as a reaction intermediate (Fig. 13-8d). With departure of the leaving group (ADP), the transfer of a phosphoryl group is complete. The large family of enzymes that catalyze phosphoryl group transfers with ATP as donor are called kinases (Greek kinein, “to move”). Hexokinase, for example, “moves” a phosphoryl group from ATP to the hexose glucose. Box 13-1 offers a primer on some of the broad classes of enzymes (including kinases) that you will encounter in your study of metabolism.
FIGURE 13-8 Phosphoryl group transfers: some of the participants. (a) In one (inadequate) representation of , three oxygens are single-bonded to phosphorus, and the fourth is double-bonded, allowing the four different resonance structures shown here. (b) The resonance structures of can be represented more accurately by showing all four phosphorus–oxygen bonds with some double-bond character; the hybrid orbitals so represented are arranged in a tetrahedron with P at its center. (c) When a nucleophile Z (in this case, the on C-6 of glucose) attacks ATP, it displaces ADP (W). In this reaction, a pentacovalent intermediate (d) forms transiently.
Phosphoryl groups are not the only groups that activate molecules for reaction. Thioalcohols (thiols), in which the oxygen atom of an alcohol is replaced with a sulfur atom, are also good leaving groups. Thiols activate carboxylic acids by forming thioesters (thiol esters). In later chapters we discuss several reactions, including those catalyzed by the fatty acyl synthases in lipid synthesis (see Fig. 21-2), in which nucleophilic substitution at the carbonyl carbon of a thioester results in transfer of the acyl group to another moiety.
Oxidation-Reduction Reactions
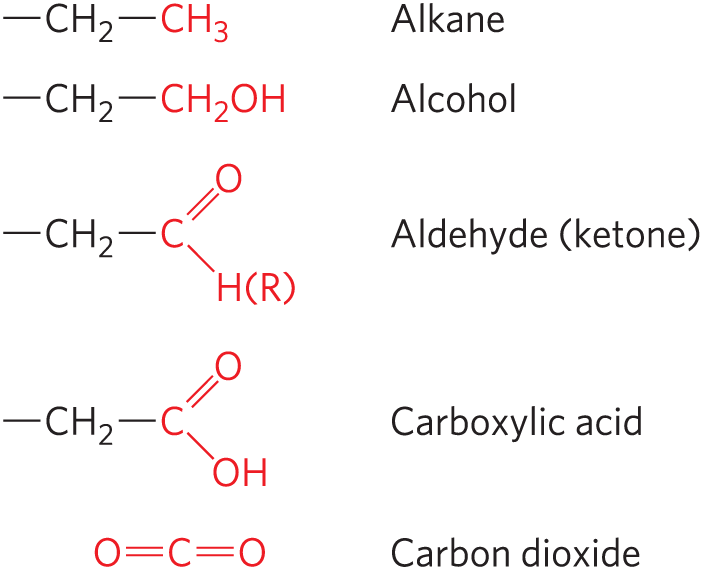
We will encounter carbon atoms in five oxidation states, depending on the elements with which they share electrons (Fig. 13-9), and transitions between these states are of crucial importance in metabolism (oxidation-reduction reactions are the topic of Section 13.4). In many biological oxidations, a compound loses two electrons and two hydrogen ions (that is, two hydrogen atoms); these reactions are commonly called dehydrogenations, and the enzymes that catalyze them are called dehydrogenases (Fig. 13-10). In some, but not all, biological oxidations, a carbon atom becomes covalently bonded to an oxygen atom. The enzymes that catalyze oxidations with oxygen as electron acceptor are generally called oxidases or, if the oxygen atom is derived directly from molecular oxygen and incorporated into the product, oxygenases.
FIGURE 13-9 The oxidation levels of carbon in biomolecules. Each compound is formed by oxidation of the carbon shown in red in the compound immediately above. Carbon dioxide is the most highly oxidized form of carbon found in living systems.
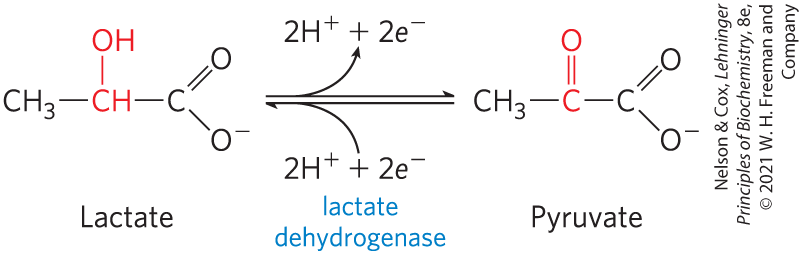
FIGURE 13-10 An oxidation-reduction reaction. Shown here is the oxidation of lactate to pyruvate. In this dehydrogenation, two electrons and two hydrogen ions (the equivalent of two hydrogen atoms) are removed from C-2 of lactate, an alcohol, to form pyruvate, a ketone. In cells the reaction is catalyzed by lactate dehydrogenase and the electrons are transferred to the cofactor nicotinamide adenine dinucleotide . This reaction is fully reversible; pyruvate can be reduced by electrons transferred from the cofactor.
Every oxidation must be accompanied by a reduction, in which an electron acceptor acquires the electrons removed by oxidation. Oxidation reactions generally release energy (think of campfires: the compounds in wood are oxidized by oxygen molecules in the air). Most living cells obtain the energy needed for cellular work by oxidizing metabolic fuels such as carbohydrates or fat (photosynthetic organisms can also trap and use the energy of sunlight). The catabolic (energy-yielding) pathways described in Chapters 14 through 19 are oxidative reaction sequences that result in the transfer of electrons from fuel molecules, through a series of electron carriers, to oxygen. The high affinity of for electrons makes the overall electron-transfer process highly exergonic, providing the energy that drives ATP synthesis — the central goal of catabolism.
Many of the reactions within these five classes are facilitated by cofactors, in the form of coenzymes and metal ions (, S-adenosylmethionine, folate, nicotinamide, and are some examples). Cofactors bind to enzymes — in some cases reversibly, in other cases almost irreversibly — and give them the capacity to promote a particular kind of chemistry (p. 178). Most cofactors participate in a narrow range of closely related reactions. In the following chapters, we introduce and discuss each important cofactor at the point where we first encounter its function. The cofactors provide another way to organize the study of biochemical processes, given that the reactions facilitated by a given cofactor generally are mechanistically related.
Biochemical and Chemical Equations Are Not Identical
Biochemists write metabolic equations in a simplified way, and this is particularly evident for reactions involving ATP. Phosphorylated compounds can exist in several ionization states and, as we have noted, the different species can bind . For example, at pH 7 and 2 mm , ATP exists as an equilibrium distribution of the forms , , , , and . In thinking about the biological role of ATP, however, we are not always interested in all this detail, and so we consider ATP as an entity made up of a sum of species, and we write its hydrolysis as the biochemical equation
where ATP, ADP, and are sums of species. The corresponding standard transformed equilibrium constant, , depends on the pH and the concentration of free . Note that and do not appear in the biochemical equation, because their concentrations are not significantly changed by the reaction. Thus a biochemical equation does not necessarily balance H, Mg, or charge, although it does balance all other elements involved in the reaction (C, N, O, and P in the equation above).
We can write a chemical equation that does balance for all elements and for charge. For example, when ATP is hydrolyzed at a pH above 8.5 in the absence of , the chemical reaction is represented by
The corresponding equilibrium constant,
depends only on temperature, pressure, and ionic strength.
Both ways of writing a metabolic reaction have value in biochemistry. Chemical equations are needed when we want to account for all atoms and charges in a reaction, as when we are considering the mechanism of a chemical reaction. Biochemical equations are used to determine in which direction a reaction will proceed spontaneously, given a specified pH and , or to calculate the equilibrium constant of such a reaction.
Throughout this book we use biochemical equations, unless the focus is on chemical mechanism, and we use values of and as determined at pH 7 and 1 mm.
SUMMARY 13.2 Chemical Logic and Common Biochemical Reactions
Living systems make use of a large number of chemical reactions that can be classified into five general types: reactions that make or break carbon–carbon bonds; internal rearrangements and eliminations; free-radical reactions; group transfers; and oxidation-reduction reactions. Heterolytic cleavages occur often in reactions that make or break bonds.
Carbonyl groups play a special role in reactions that form or cleave bonds. Carbanion intermediates are common and are stabilized by adjacent carbonyl groups or, less often, by imines or certain cofactors.
A redistribution of electrons can produce internal rearrangements, isomerizations, and eliminations. Such reactions include intramolecular oxidation-reduction, change in cis-trans arrangement at a double bond, and transposition of double bonds.
Homolytic cleavage of covalent bonds to generate free radicals occurs in some pathways.
Phosphoryl transfer reactions are an especially important type of group transfer in cells, required for the activation of molecules for reactions that would otherwise be highly unfavorable.
Oxidation-reduction reactions involve the loss or gain of electrons: one reactant gains electrons and is reduced, while the other loses electrons and is oxidized. Oxidation reactions generally release energy and are important in catabolism.
Biochemists often write reaction equations that are not balanced for and don’t attempt to describe the state of phosphate ionization.
 There are patterns within the chemistry of life; you do not need to learn every individual reaction to comprehend the molecular logic of biochemistry. Most of the reactions in living cells fall into one of five general categories: (1) reactions that make or break carbon–carbon bonds; (2) internal rearrangements, isomerizations, and eliminations; (3) free-radical reactions; (4) group transfers; and (5) oxidation-reductions. We discuss each of these in more detail below and refer to some examples of each type in later chapters. Note that the five reaction types are not mutually exclusive; for example, an isomerization reaction may involve a free-radical intermediate.
There are patterns within the chemistry of life; you do not need to learn every individual reaction to comprehend the molecular logic of biochemistry. Most of the reactions in living cells fall into one of five general categories: (1) reactions that make or break carbon–carbon bonds; (2) internal rearrangements, isomerizations, and eliminations; (3) free-radical reactions; (4) group transfers; and (5) oxidation-reductions. We discuss each of these in more detail below and refer to some examples of each type in later chapters. Note that the five reaction types are not mutually exclusive; for example, an isomerization reaction may involve a free-radical intermediate.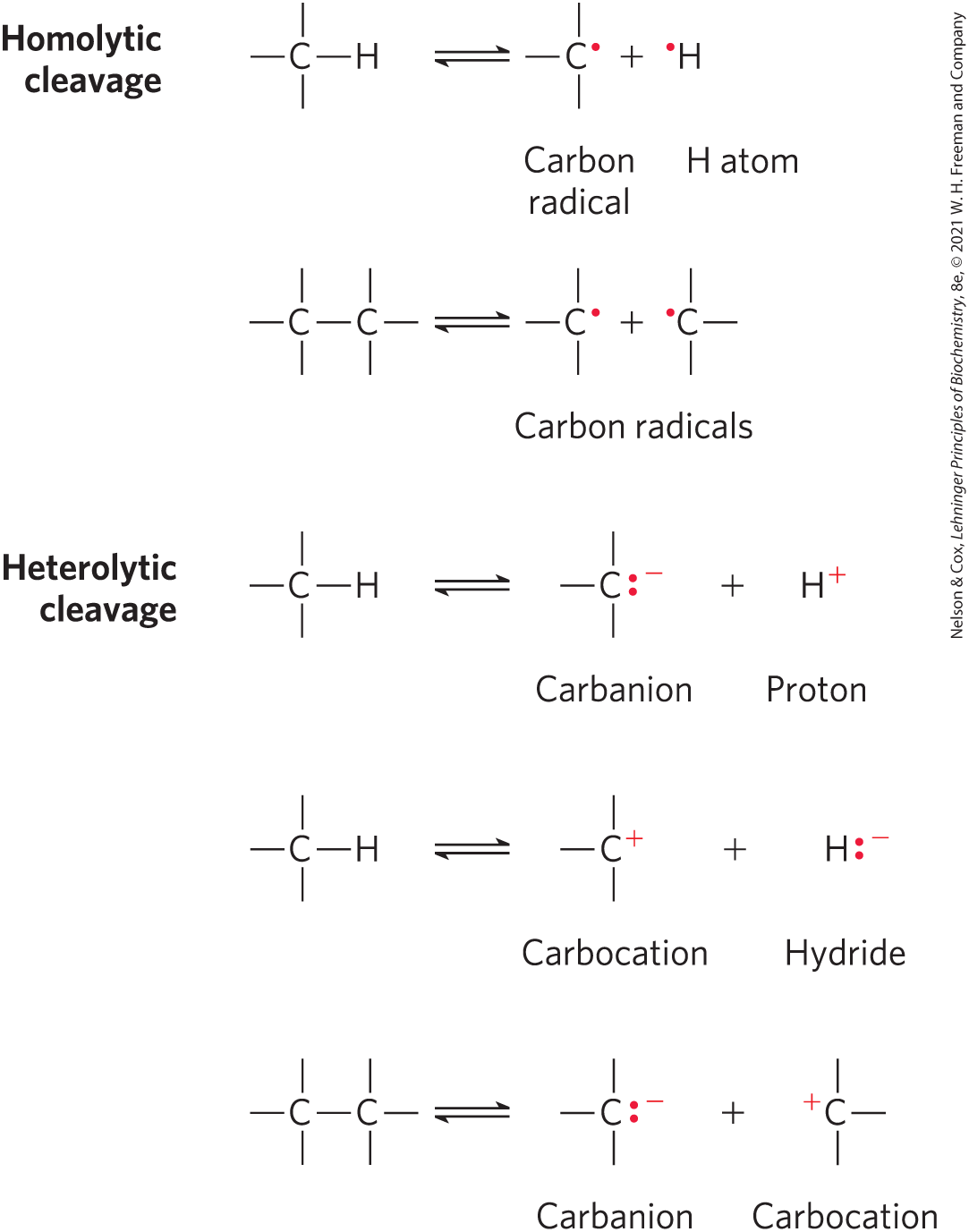
 ) represent the movement of electron pairs. For movement of a single electron (as in a free-radical reaction), a single-headed (fishhook-type) arrow is used (
) represent the movement of electron pairs. For movement of a single electron (as in a free-radical reaction), a single-headed (fishhook-type) arrow is used (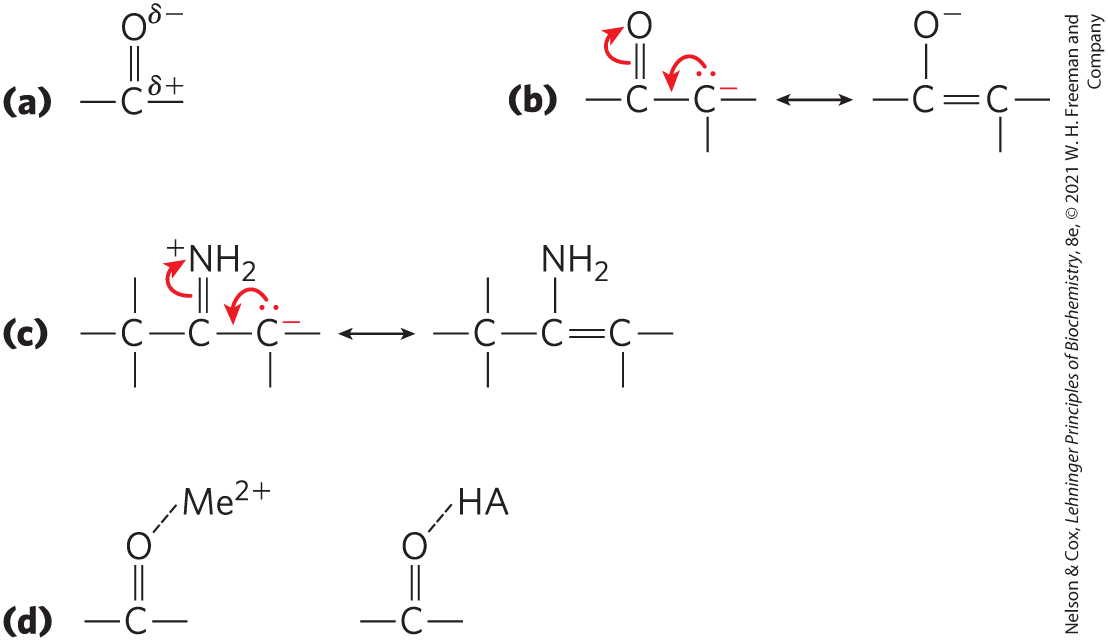
 Most living cells obtain the energy needed for cellular work by oxidizing metabolic fuels such as carbohydrates or fat (photosynthetic organisms can also trap and use the energy of sunlight). The catabolic (energy-yielding) pathways described in
Most living cells obtain the energy needed for cellular work by oxidizing metabolic fuels such as carbohydrates or fat (photosynthetic organisms can also trap and use the energy of sunlight). The catabolic (energy-yielding) pathways described in  Living systems make use of a large number of chemical reactions that can be classified into five general types: reactions that make or break carbon–carbon bonds; internal rearrangements and eliminations; free-radical reactions; group transfers; and oxidation-reduction reactions. Heterolytic cleavages occur often in reactions that make or break bonds.
Living systems make use of a large number of chemical reactions that can be classified into five general types: reactions that make or break carbon–carbon bonds; internal rearrangements and eliminations; free-radical reactions; group transfers; and oxidation-reduction reactions. Heterolytic cleavages occur often in reactions that make or break bonds.