Nitrogen, , is abundant in the atmosphere but is too unreactive for use in most biochemical processes. Reduced nitrogen is essential for life but bioenergetically expensive. Only a few microorganisms can convert to biologically useful forms such as (Chapter 22). Amino groups are thus used efficiently in biological systems, and the lack of reactive nitrogen can limit growth.
If not reused for the synthesis of new amino acids or other nitrogenous products, amino groups are channeled into a single excretory end product (Fig. 18-2). Most aquatic species, such as the bony fishes, are ammonotelic, excreting amino nitrogen as ammonia. The toxic ammonia is simply diluted in the surrounding water. Terrestrial animals require pathways for nitrogen excretion that minimize toxicity and water loss. Most terrestrial animals are ureotelic, excreting amino nitrogen in the form of urea; birds and reptiles are uricotelic, excreting amino nitrogen as uric acid. (The pathway of uric acid synthesis is described in Fig. 22-48.) Plants recycle virtually all amino groups — they excrete nitrogen only under rare circumstances. Reactive nitrogen excreted in any form is rapidly assimilated into the global nitrogen web (see Fig. 22-1), metabolized by microorganisms that are ubiquitous in aqueous and soil environments.
FIGURE 18-2 Amino group catabolism. (a) Overview of catabolism of amino groups (shaded) in vertebrate liver. (b) Excretory forms of nitrogen. Excess is excreted as ammonia, urea, or uric acid. Notice that the carbon atoms of urea and uric acid are highly oxidized; the organism discards carbon only after extracting most of its available energy of oxidation.
Figure 18-2a provides an overview of the catabolic pathways of ammonia and amino groups in vertebrates. Glutamine, glutamate, and alanine are prominent in this scheme. Amino acids derived from dietary protein are the source of most amino groups. Most amino acids are metabolized in the liver. Some of the ammonia generated in this process is recycled and used in biosynthetic pathways where glutamine, glutamate, and aspartate play major roles (Chapter 22). Excess amino groups are either excreted directly or converted to urea or uric acid for excretion, depending on the organism (Fig. 18-2b). In mammals, including marsupials, most excess ammonia generated in other (extrahepatic) tissues travels to the liver for conversion to urea.
The special place of glutamate, glutamine, alanine, and aspartate in nitrogen metabolism is not an evolutionary accident. These particular amino acids are the ones most easily converted into citric acid cycle intermediates: glutamate and glutamine to α-ketoglutarate, alanine to pyruvate, and aspartate to oxaloacetate. Glutamate and glutamine are especially important, acting as general collection points for amino groups. In the cytosol of liver cells (hepatocytes), amino groups from most amino acids are transferred to α-ketoglutarate to form glutamate, which enters mitochondria and gives up its amino group to form . Excess ammonia generated in most other tissues is converted to the amide nitrogen of glutamine, which passes to the liver, then into liver mitochondria. Glutamine or glutamate or both are present in higher concentrations than other amino acids in most tissues. In skeletal muscle, excess amino groups are generally transferred to pyruvate to form alanine, another important molecule in the transport of amino groups to the liver. We begin with a discussion of the breakdown of dietary proteins, then proceed to a general description of the metabolic fates of amino groups.
Dietary Protein Is Enzymatically Degraded to Amino Acids
In humans, the degradation of ingested proteins to their constituent amino acids occurs in the gastrointestinal tract. Entry of dietary protein into the stomach stimulates the gastric mucosa to secrete the hormone gastrin, which in turn stimulates the secretion of hydrochloric acid by the parietal cells and pepsinogen by the chief cells of the gastric glands (Fig. 18-3a). The acidic gastric juice (pH 1.0 to 2.5) is both an antiseptic, killing most bacteria and other foreign cells, and a denaturing agent, unfolding globular proteins and rendering their internal peptide bonds more accessible to enzymatic hydrolysis. Pepsinogen , an inactive precursor, or zymogen (p. 220), is converted to active pepsin by an autocatalytic cleavage (a cleavage mediated by the pepsinogen itself) that occurs only at low pH. In the stomach, pepsin cleaves long polypeptide chains into a mixture of smaller peptides.
FIGURE 18-3 Part of the human digestive (gastrointestinal) tract. (a) The parietal cells and chief cells of the gastric glands secrete their products in response to the hormone gastrin. Pepsin begins the process of protein degradation in the stomach. (b) The cytoplasm of exocrine cells of the pancreas is completely filled with rough endoplasmic reticulum, the site of synthesis of the zymogens of many digestive enzymes. The zymogens are concentrated in membrane-enclosed transport particles called zymogen granules. When an exocrine cell is stimulated, its plasma membrane fuses with the zymogen granule membrane and zymogens are released into the lumen of the collecting duct by exocytosis. The collecting ducts ultimately lead to the pancreatic duct and from there to the small intestine. (c) In the small intestine, amino acids are absorbed through the epithelial cell layer (intestinal mucosa) of the villi and enter the capillaries.
As the acidic stomach contents pass into the small intestine, the low pH triggers secretion of the hormone secretin into the blood. Secretin stimulates the pancreas to secrete bicarbonate into the small intestine to neutralize the gastric HCl, abruptly increasing the pH to about 7. Arrival of peptides in the upper part of the intestine (duodenum) causes release into the blood of the hormone cholecystokinin, which stimulates secretion of several pancreatic proteases with activity optima at pH 7 to 8. Trypsinogen, chymotrypsinogen, and procarboxypeptidases A and B—the zymogens of trypsin, chymotrypsin, and carboxypeptidases A and B—are synthesized and secreted by the exocrine cells of the pancreas (Fig. 18-3b). Trypsinogen is converted to its active form, trypsin, by enteropeptidase, a proteolytic enzyme secreted by intestinal cells. Free trypsin then catalyzes the conversion of additional trypsinogen to trypsin (see Fig. 6-42). Trypsin also activates chymotrypsinogen, the procarboxypeptidases, and proelastase.
Why this elaborate mechanism for getting active digestive enzymes into the gastrointestinal tract? Synthesis of the enzymes as inactive precursors protects the exocrine cells from destructive proteolytic attack. The pancreas further protects itself against self-digestion by making a specific inhibitor, a protein called pancreatic trypsin inhibitor (p. 220). Given the key role of trypsin in proteolytic activation pathways, inhibition of trypsin effectively prevents premature production of active proteolytic enzymes within pancreatic cells.
Trypsin and chymotrypsin further hydrolyze the peptides that were produced by pepsin in the stomach. This stage of protein digestion is accomplished very efficiently, because pepsin, trypsin, chymotrypsin, and the carboxypeptidases have different catalytic specificities and cleave different sets of peptide bonds (see Table 3-6). The resulting mixture of free amino acids is transported into the epithelial cells lining the small intestine (Fig. 18-3c), through which the amino acids enter the blood capillaries in the villi and travel to the liver.
Acute pancreatitis is a disease caused by obstruction of the normal pathway by which pancreatic secretions enter the intestine. The zymogens of the proteolytic enzymes are converted to their catalytically active forms prematurely, inside the pancreatic cells, and attack the pancreatic tissue itself. This causes excruciating pain and damage to the organ that can prove fatal.
Pyridoxal Phosphate Participates in the Transfer of α-Amino Groups to α-Ketoglutarate
The first step in the catabolism of most l-amino acids, once they have reached the liver, is removal of the α-amino groups, promoted by enzymes called aminotransferases or transaminases. In these transamination reactions, the α-amino group is transferred to the α-carbon atom of α-ketoglutarate, leaving behind the corresponding α-keto acid analog of the amino acid (Fig. 18-4). There is no net deamination (loss of amino groups) in these reactions, because the α-ketoglutarate becomes aminated as the α-amino acid is deaminated. These are the reactions that effectively collect the amino groups from many different amino acids in the form of l-glutamate.
FIGURE 18-4 Enzyme-catalyzed transaminations. In many aminotransferase reactions, α-ketoglutarate is the amino group acceptor. All aminotransferases have pyridoxal phosphate (PLP) as cofactor. Although the reaction is shown here in the direction of transfer of the amino group to α-ketoglutarate, it is readily reversible.
Cells contain different types of aminotransferases. Many are specific for α-ketoglutarate as the amino group acceptor but differ in their specificity for the l-amino acid. The enzymes are named for the amino group donor (alanine aminotransferase and aspartate aminotransferase, for example). The reactions catalyzed by aminotransferases are freely reversible, having an equilibrium constant of about .
All aminotransferases have pyridoxal phosphate (PLP), the coenzyme form of pyridoxine, or vitamin , as a prosthetic group. We encountered pyridoxal phosphate in Chapter 15, as a coenzyme in the glycogen phosphorylase reaction, but its role in that reaction is not representative of its usual coenzyme function. Its primary role in cells is in the metabolism of molecules with amino groups.
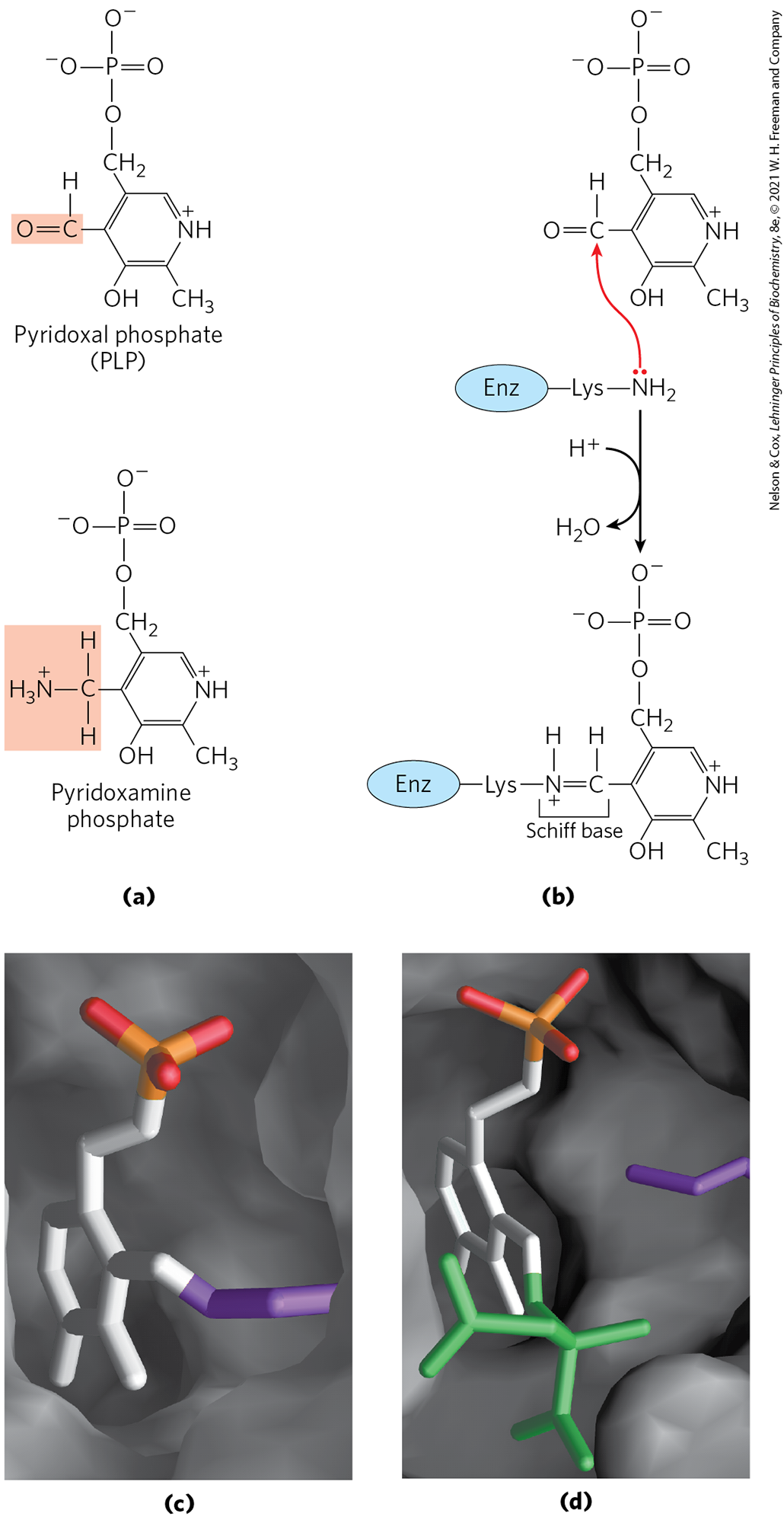
Pyridoxal phosphate functions as an intermediate carrier of amino groups at the active site of aminotransferases. It undergoes reversible transformations between its aldehyde form, pyridoxal phosphate, which can accept an amino group, and its aminated form, pyridoxamine phosphate, which can donate its amino group to an α-keto acid (Fig. 18-5a). Pyridoxal phosphate is generally covalently bound to the enzyme’s active site through an aldimine (Schiff base) linkage to the ε-amino group of a Lys residue (Fig. 18-5b, c). This linkage is replaced by the amino group of the amino acid as the first step in most PLP-catalyzed reactions (Fig. 18-5d).
FIGURE 18-5 Pyridoxal phosphate, the prosthetic group of aminotransferases. (a) Pyridoxal phosphate (PLP) and its aminated form, pyridoxamine phosphate, are the tightly bound coenzymes of aminotransferases. The functional groups are shaded. (b) Pyridoxal phosphate is bound to the enzyme through noncovalent interactions and a Schiff-base (aldimine) linkage to a Lys residue at the active site. The steps in the formation of a Schiff base from a primary amine and a carbonyl group are detailed in Figure 14-5. (c, d) Close-up views of the active site of aspartate aminotransferase, with PLP (white, with the phosphoryl group in orange and red). In (c) PLP is in aldimine linkage with the side chain of (purple). In (d) PLP is linked to the substrate analog 2-methylaspartate (green) via a Schiff base. [(c, d) Data from PDB ID 1AJS, S. Rhee et al., J. Biol. Chem. 272:17,293, 1997.]
Pyridoxal phosphate participates in a variety of reactions in amino acid metabolism (Section 18.3 and Chapter 22), facilitating reactions at the α, β, and γ carbons (C-2 to C-4) of amino acids. Reactions at the α carbon (Fig. 18-6) include racemizations (interconverting l- and d-amino acids) and decarboxylations, as well as transaminations. Pyridoxal phosphate plays the same chemical role in each of these reactions. A bond to the α carbon of the substrate is broken, removing either a proton or a carboxyl group. Pyridoxal phosphate provides resonance stabilization of the otherwise unstable carbanion intermediate (Fig. 18-6, inset). The highly conjugated structure of PLP (an electron sink) permits delocalization of the negative charge.
MECHANISM FIGURE 18-6 Some amino acid transformations at the α carbon that are facilitated by pyridoxal phosphate. Pyridoxal phosphate is generally bonded to the enzyme through a Schiff base, also called an internal aldimine. This activated form of PLP readily undergoes transamination to form a new Schiff base (external aldimine) with the α-amino group of the substrate amino acid (see Fig. 18-5b, d). Three alternative fates for the external aldimine are shown: transamination, racemization, and decarboxylation. The PLP–amino acid Schiff base is in conjugation with the pyridine ring, an electron sink that permits delocalization of an electron pair to avoid formation of an unstable carbanion on the α carbon (inset). A quinonoid intermediate is involved in all three types of reactions. The transamination route is especially important in the pathways described in this chapter. This pathway (shown left to right) represents only part of the overall reaction catalyzed by aminotransferases. To complete the process, a second α-keto acid replaces the one that is released, and this is converted to an amino acid in a reversal of the reaction steps (right to left). Pyridoxal phosphate is also involved in certain reactions at the β and γ carbons of some amino acids (not shown).
Aminotransferases (Fig. 18-6a) are classic examples of enzymes catalyzing bimolecular Ping-Pong reactions (see Fig. 6-15b, d), in which the first substrate reacts and the product must leave the active site before the second substrate can bind. Thus, the incoming amino acid binds to the active site, donates its amino group to pyridoxal phosphate, and departs in the form of an α-keto acid. The incoming α-keto acid then binds, accepts the amino group from pyridoxamine phosphate, and departs in the form of an amino acid.
Glutamate Releases Its Amino Group as Ammonia in the Liver
As we shall see, the urea cycle begins with free ammonia in the mitochondria of hepatocytes. The delivery of to these mitochondria is streamlined by collecting the amino groups of many different α-amino acids in the liver in one of two forms: the amino group of l-glutamate or the amide nitrogen of glutamine (Fig. 18-2).
As the product of many aminotransferase reactions, glutamate has a central role. In hepatocytes, glutamate is transported from the cytosol into mitochondria. Here, it undergoes oxidative deamination catalyzed by l-glutamate dehydrogenase to produce and α-ketoglutarate. In mammals, this enzyme is present in the mitochondrial matrix. It is unusual in that it can use either or as the acceptor of reducing equivalents (Fig. 18-7).
FIGURE 18-7 Reaction catalyzed by glutamate dehydrogenase. The glutamate dehydrogenase of mammalian liver has the unusual capacity to use either or as cofactor. The glutamate dehydrogenases of plants and microorganisms are generally specific for one or the other. The mammalian enzyme is allosterically regulated by GTP and ADP.
The combined action of an aminotransferase and glutamate dehydrogenase is referred to as transdeamination. A few amino acids bypass the transdeamination pathway and undergo direct oxidative deamination. The fate of the produced by any of these deamination processes is discussed in detail in Section 18.2. The α-ketoglutarate formed from glutamate deamination can be used either in the citric acid cycle or for glucose synthesis.
Glutamate dehydrogenase operates at an important intersection of carbon and nitrogen metabolism. Its α-ketoglutarate product can be oxidized as fuel or serve as a glucose precursor in gluconeogenesis. An allosteric enzyme with six identical subunits, its activity is influenced by a complicated array of allosteric modulators. The best-studied of these are the positive modulator ADP and the negative modulator GTP. Although the metabolic rationale for this regulatory pattern has not been elucidated in detail, ADP can signal low glucose levels and GTP is a product of the citric acid cycle that can signal high levels of α-ketoglutarate (see Fig. 16-7).
Mutations that alter the allosteric binding site for GTP or otherwise cause permanent activation of glutamate dehydrogenase lead to a human genetic disorder called hyperinsulinism-hyperammonemia syndrome, characterized by hypersecretion of insulin after a protein meal. This results in elevated levels of ammonia in the bloodstream and hypoglycemia.
Glutamine Transports Ammonia in the Bloodstream
Glutamine is the second major source of ammonia in hepatocyte mitochondria and is particularly important for intercellular transport of ammonia. Ammonia is quite toxic to animal tissues (we examine some possible reasons for this toxicity later). Significant amounts are present in blood, but the levels are tightly controlled. In many tissues, including the brain, some processes such as nucleotide degradation generate free ammonia. In most animals, much of the free ammonia is converted to a nontoxic compound before export from the extrahepatic tissues into the blood and transport to the liver or kidneys. For this transport function, glutamate, critical to intracellular amino group metabolism, is supplanted by l-glutamine. The free ammonia produced in tissues is combined with glutamate to yield glutamine by the action of glutamine synthetase. This reaction requires ATP and occurs in two steps (Fig. 18-8). First, glutamate and ATP react to form ADP and a γ-glutamyl phosphate intermediate, which then reacts with ammonia to produce glutamine and inorganic phosphate. In addition to its transport role, glutamine serves as a source of amino groups in a variety of biosynthetic reactions. Glutamine synthetase is found in all organisms, always playing a central metabolic role. In microorganisms, the enzyme serves as an essential portal for the entry of fixed nitrogen into biological systems. (The roles of glutamine and glutamine synthetase in metabolism are further discussed in Chapter 22.)
FIGURE 18-8 Ammonia transport in the form of glutamine. Excess ammonia in tissues is added to glutamate to form glutamine, a process catalyzed by glutamine synthetase. After transport in the bloodstream, the glutamine enters the liver, and is liberated in mitochondria by the enzyme glutaminase.
In most terrestrial animals, glutamine in excess of that required for biosynthesis is transported in the blood to the intestine, liver, and kidneys for processing. In these tissues, the amide nitrogen is released as ammonium ion in the mitochondria, where the enzyme glutaminase converts glutamine to glutamate and (Fig. 18-8). The from intestine and kidney is transported in the blood to the liver. In the liver, the ammonia from all sources is disposed of by urea synthesis. Some of the glutamate produced in the glutaminase reaction may be further processed in the liver by glutamate dehydrogenase (Fig. 18-7), releasing more ammonia and producing carbon skeletons for metabolic fuel.
In metabolic acidosis (p. 621) there is a regulated increase in glutamine processing by the kidneys. Much of the excess thus produced is not destined for the bloodstream or converted to urea; instead, it is excreted directly into the urine where it forms salts with metabolic acids. The glutamine breakdown thus facilitates removal of those acids in the urine. Bicarbonate produced by the decarboxylation of α-ketoglutarate in the citric acid cycle can also serve as a buffer in blood plasma. Taken together, these effects of glutamine metabolism in the kidney tend to counteract acidosis.
Alanine Transports Ammonia from Skeletal Muscles to the Liver
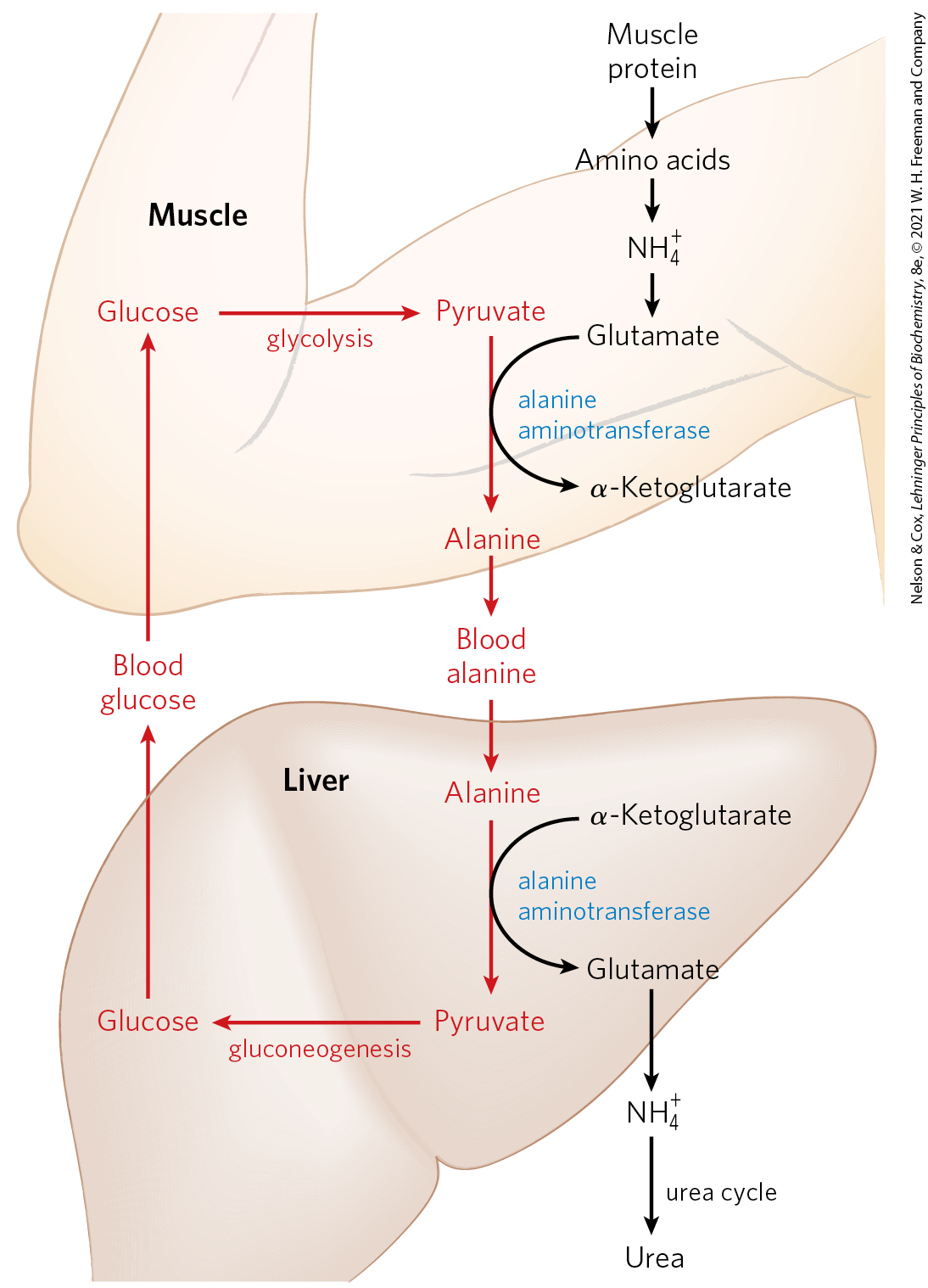
Vigorously contracting skeletal muscles operate anaerobically, producing large amounts of pyruvate and lactate from glycolysis as well as ammonia from protein breakdown. These products must find their way to the liver, where pyruvate and lactate are incorporated into glucose, which is returned to the muscles, and ammonia is converted to urea for excretion. Pyruvate and alanine are readily interconverted via transamination with glutamate by the action of alanine aminotransferase). Thus, alanine largely supplants glutamine in the transport of amino groups from muscle to the liver in a nontoxic form (Fig. 18-2a), ultimately delivering the free ammonia to hepatocyte mitochondria via glutamate in a pathway called the glucose-alanine cycle (Fig. 18-9). In the cytosol of hepatocytes, alanine aminotransferase acts in reverse to transfer the amino group from alanine to α-ketoglutarate, forming pyruvate and glutamate. Glutamate can then enter mitochondria, where the glutamate dehydrogenase reaction releases (Fig. 18-7), or it can undergo transamination with oxaloacetate to form aspartate, another nitrogen donor in urea synthesis, as we shall see.
FIGURE 18-9 Glucose-alanine cycle. Alanine serves as a carrier of ammonia and of the carbon skeleton of pyruvate from skeletal muscle to liver. The ammonia is excreted and the pyruvate is used to produce glucose, which is returned to the muscle.
The use of alanine to transport ammonia from skeletal muscles to the liver is another example of the intrinsic economy of living organisms. The energetic burden of gluconeogenesis is imposed on the liver rather than the muscle, and all available ATP in muscle is devoted to muscle contraction. The glucose-alanine cycle, in concert with the Cori cycle (see Box 14-2 and Fig. 23-17), accomplishes this transaction.
Ammonia Is Toxic to Animals
The catabolic production of ammonia poses a serious biochemical problem, because ammonia is very toxic. The brain is particularly sensitive; damage from ammonia toxicity causes cognitive impairment, ataxia, and epileptic seizures. In extreme cases there is swelling of the brain leading to death. The molecular bases for this toxicity are gradually coming into focus. In the blood, about 98% of ammonia is in the protonated form , which does not cross the plasma membrane. The small amount of present readily crosses all membranes, including the blood-brain barrier, allowing it to enter cells, where much of it becomes protonated and can accumulate inside cells as .
Ridding the cytosol of ammonia requires reductive amination of α-ketoglutarate to glutamate by glutamate dehydrogenase (the reverse of the reaction described earlier; Fig. 18-7) and conversion of glutamate to glutamine by glutamine synthetase. In the brain, only astrocytes — star-shaped cells of the nervous system that provide nutrients, support, and insulation for neurons — express glutamine synthetase. Glutamate and its derivative γ-aminobutyrate (GABA; see Fig. 22-31) are important neurotransmitters; some of the sensitivity of the brain to ammonia may reflect depletion of glutamate in the glutamine synthetase reaction. However, glutamine synthetase activity is insufficient to deal with excess ammonia, or to fully explain its toxicity.
Increased also alters the capacity of astrocytes to maintain potassium homeostasis across the membrane. competes with for transport into the cell through the ATPase, resulting in elevated extracellular . The excess extracellular enters neurons through a symporter, cotransporter 1 (NKCC1), bringing and with it. Excess in these neurons alters their response when the neurotransmitter GABA interacts with their receptors, producing abnormal depolarization and increased neuronal activity that likely account for the neuromuscular incoordination and seizures that often result from ammonia poisoning. If extracellular remains elevated, the perturbation of ion and aquaporin channels in astrocytes causes the cells to swell, resulting in fatal brain edema.
As we close this discussion of amino group metabolism, note that we have described several processes that deposit excess ammonia in the mitochondria of hepatocytes (Fig. 18-2). We now look at the fate of that ammonia.
SUMMARY 18.1 Metabolic Fates of Amino Groups
Humans derive a small fraction of their oxidative energy from the catabolism of amino acids. Proteases degrade ingested proteins in the stomach and small intestine. Most proteases are initially synthesized as inactive zymogens.
An early step in the catabolism of amino acids is the separation of the amino group from the carbon skeleton. In most cases, the amino group is transferred to α-ketoglutarate to form glutamate. This transamination reaction requires pyridoxal phosphate, a coenzyme globally involved in amino acid metabolism.
Glutamate, a central reservoir of metabolized amino groups, is transported to liver mitochondria, where glutamate dehydrogenase liberates the amino group as ammonium ion .
Ammonia formed in most other tissues is transported to the liver as the amide nitrogen of glutamine.
To make use of the pyruvate and amino groups generated in hardworking skeletal muscle, the pyruvate is converted to alanine and transported to the liver within the glucose-alanine cycle.
Free ammonia is toxic. Excess ammonia is often manifested in serious neurological damage.
 If not reused for the synthesis of new amino acids or other nitrogenous products, amino groups are channeled into a single excretory end product (
If not reused for the synthesis of new amino acids or other nitrogenous products, amino groups are channeled into a single excretory end product (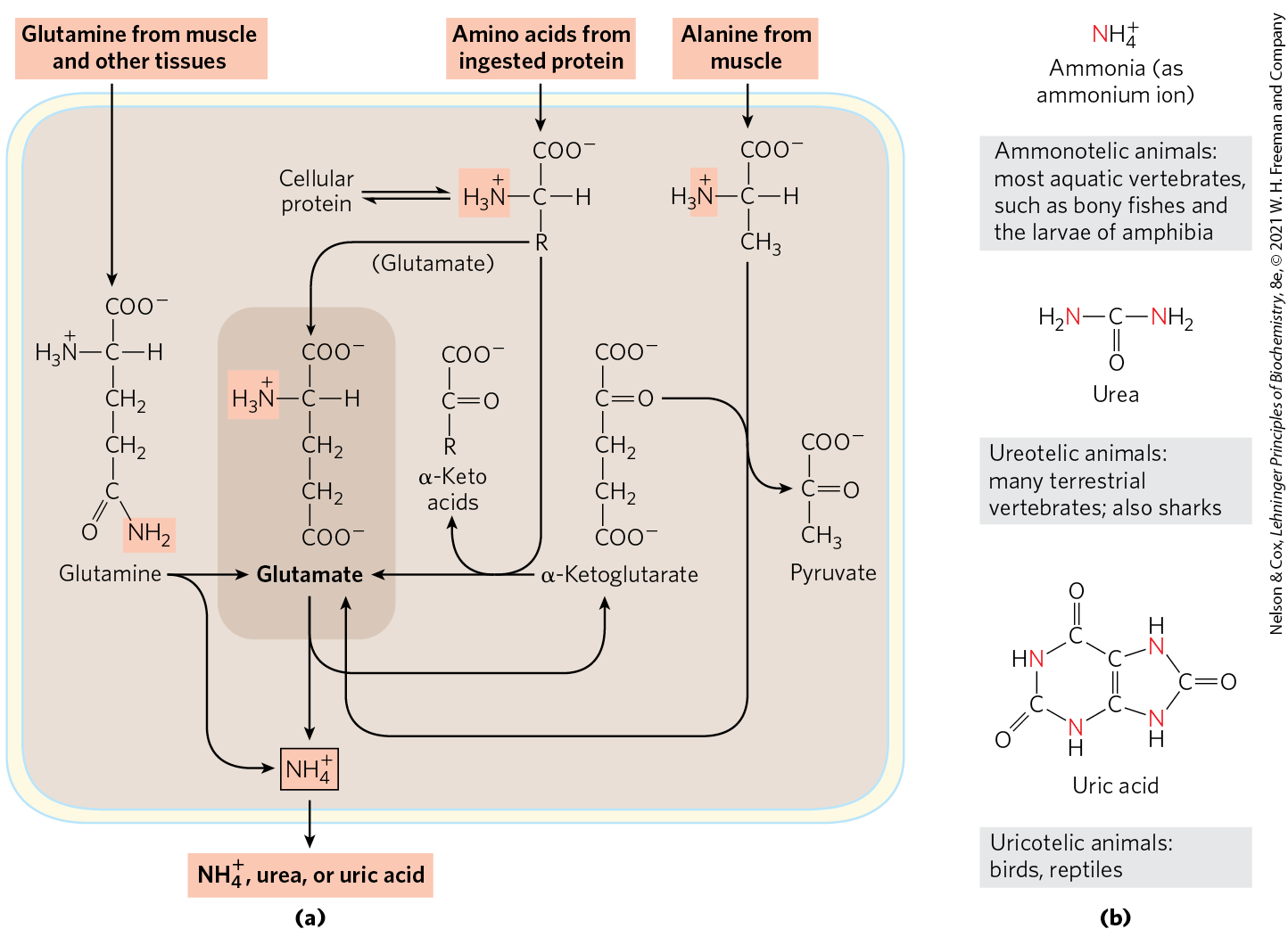
 The special place of glutamate, glutamine, alanine, and aspartate in nitrogen metabolism is not an evolutionary accident. These particular amino acids are the ones most easily converted into citric acid cycle intermediates: glutamate and glutamine to α-ketoglutarate, alanine to pyruvate, and aspartate to oxaloacetate. Glutamate and glutamine are especially important, acting as general collection points for amino groups. In the cytosol of liver cells (hepatocytes), amino groups from most amino acids are transferred to α-ketoglutarate to form glutamate, which enters mitochondria and gives up its amino group to form . Excess ammonia generated in most other tissues is converted to the amide nitrogen of glutamine, which passes to the liver, then into liver mitochondria. Glutamine or glutamate or both are present in higher concentrations than other amino acids in most tissues. In skeletal muscle, excess amino groups are generally transferred to pyruvate to form alanine, another important molecule in the transport of amino groups to the liver. We begin with a discussion of the breakdown of dietary proteins, then proceed to a general description of the metabolic fates of amino groups.
The special place of glutamate, glutamine, alanine, and aspartate in nitrogen metabolism is not an evolutionary accident. These particular amino acids are the ones most easily converted into citric acid cycle intermediates: glutamate and glutamine to α-ketoglutarate, alanine to pyruvate, and aspartate to oxaloacetate. Glutamate and glutamine are especially important, acting as general collection points for amino groups. In the cytosol of liver cells (hepatocytes), amino groups from most amino acids are transferred to α-ketoglutarate to form glutamate, which enters mitochondria and gives up its amino group to form . Excess ammonia generated in most other tissues is converted to the amide nitrogen of glutamine, which passes to the liver, then into liver mitochondria. Glutamine or glutamate or both are present in higher concentrations than other amino acids in most tissues. In skeletal muscle, excess amino groups are generally transferred to pyruvate to form alanine, another important molecule in the transport of amino groups to the liver. We begin with a discussion of the breakdown of dietary proteins, then proceed to a general description of the metabolic fates of amino groups.
 Acute pancreatitis is a disease caused by obstruction of the normal pathway by which pancreatic secretions enter the intestine. The zymogens of the proteolytic enzymes are converted to their catalytically active forms prematurely, inside the pancreatic cells, and attack the pancreatic tissue itself. This causes excruciating pain and damage to the organ that can prove fatal.
Acute pancreatitis is a disease caused by obstruction of the normal pathway by which pancreatic secretions enter the intestine. The zymogens of the proteolytic enzymes are converted to their catalytically active forms prematurely, inside the pancreatic cells, and attack the pancreatic tissue itself. This causes excruciating pain and damage to the organ that can prove fatal. 
 The first step in the catabolism of most
The first step in the catabolism of most 
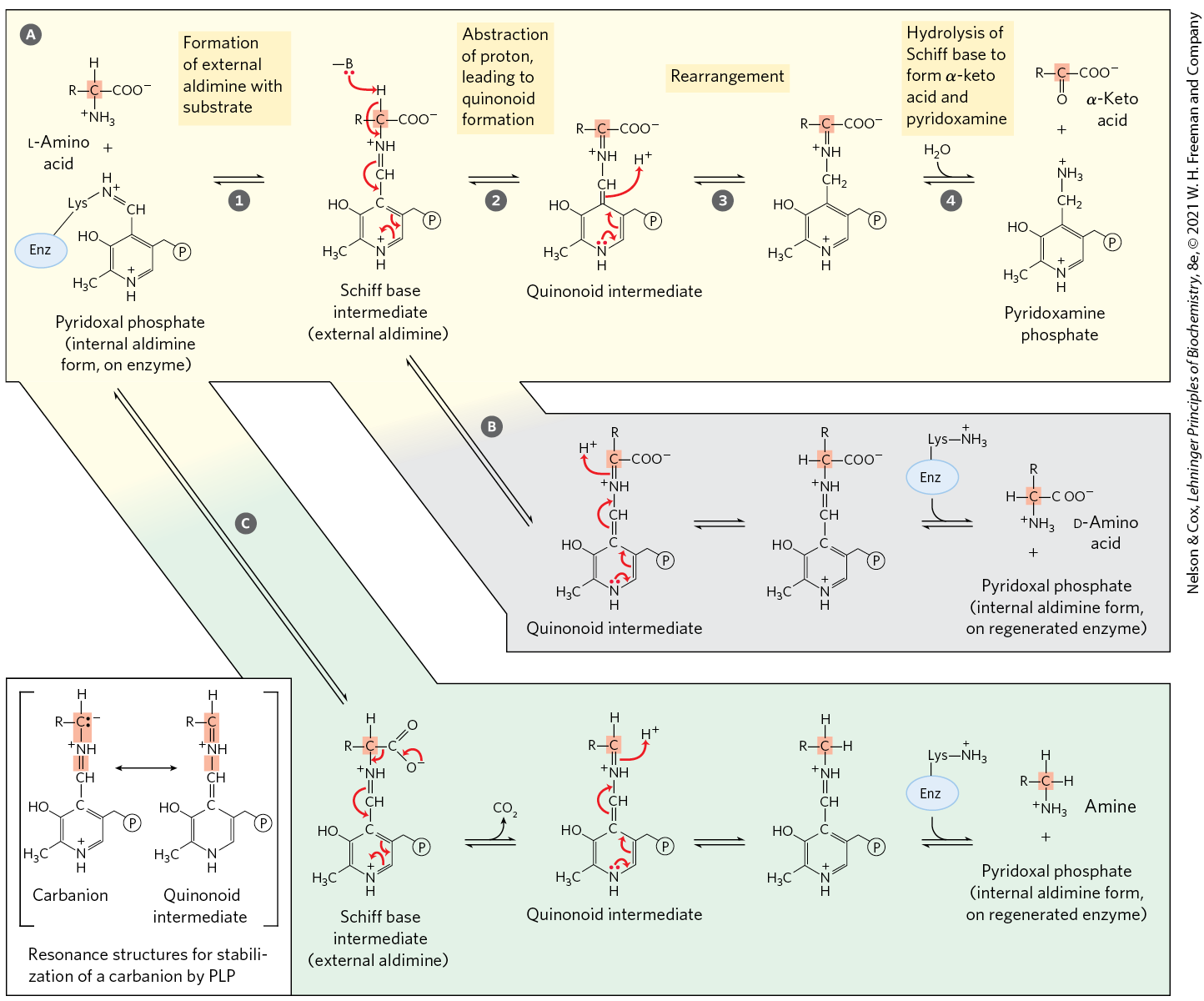
 transamination,
transamination,  racemization, and
racemization, and  decarboxylation. The PLP–amino acid Schiff base is in conjugation with the pyridine ring, an electron sink that permits delocalization of an electron pair to avoid formation of an unstable carbanion on the α carbon (inset). A quinonoid intermediate is involved in all three types of reactions. The transamination route
decarboxylation. The PLP–amino acid Schiff base is in conjugation with the pyridine ring, an electron sink that permits delocalization of an electron pair to avoid formation of an unstable carbanion on the α carbon (inset). A quinonoid intermediate is involved in all three types of reactions. The transamination route 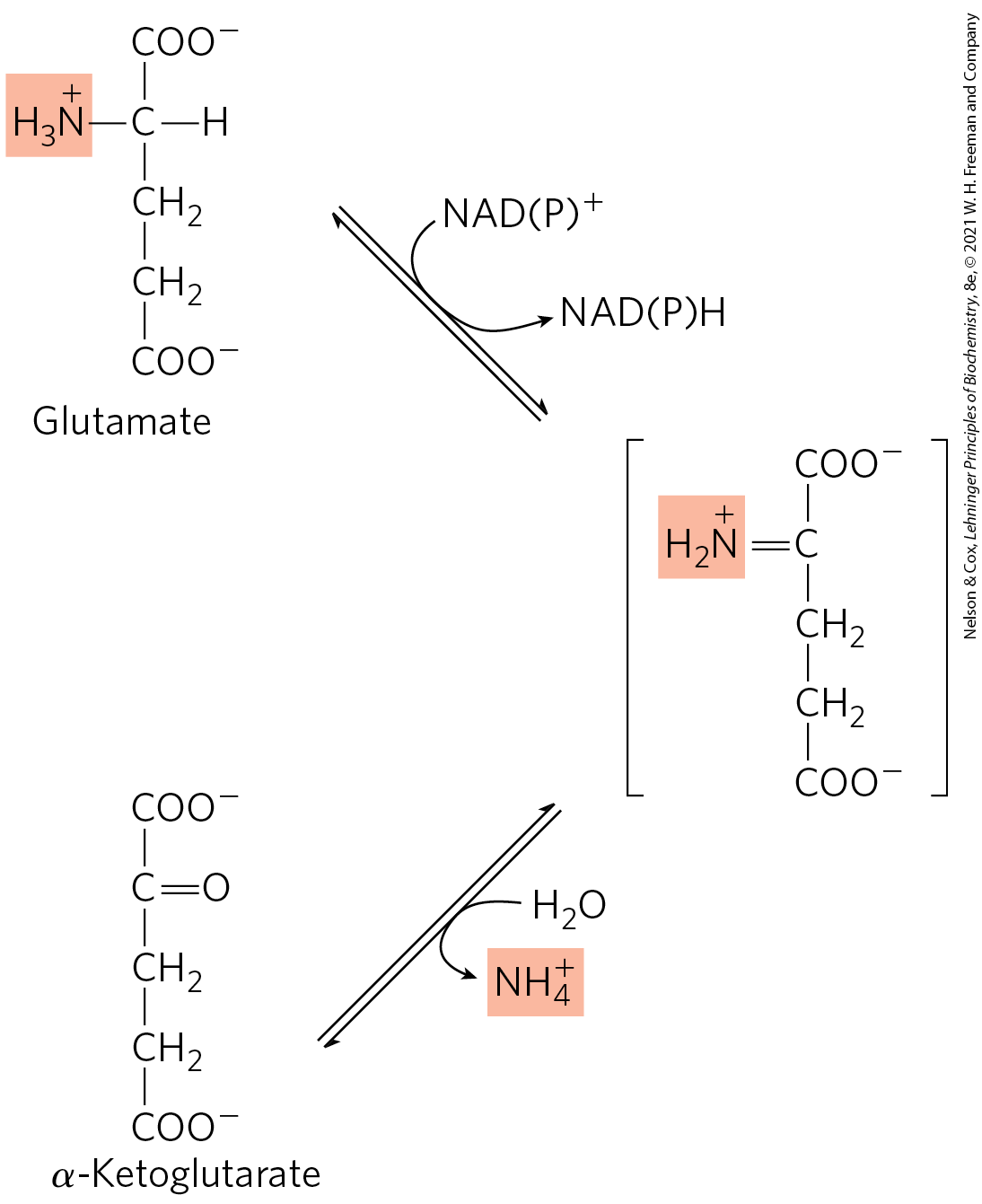
 The
The Mutations that alter the allosteric binding site for GTP or otherwise cause permanent activation of glutamate dehydrogenase lead to a human genetic disorder called hyperinsulinism-hyperammonemia syndrome, characterized by hypersecretion of insulin after a protein meal. This results in elevated levels of ammonia in the bloodstream and hypoglycemia.
Mutations that alter the allosteric binding site for GTP or otherwise cause permanent activation of glutamate dehydrogenase lead to a human genetic disorder called hyperinsulinism-hyperammonemia syndrome, characterized by hypersecretion of insulin after a protein meal. This results in elevated levels of ammonia in the bloodstream and hypoglycemia. 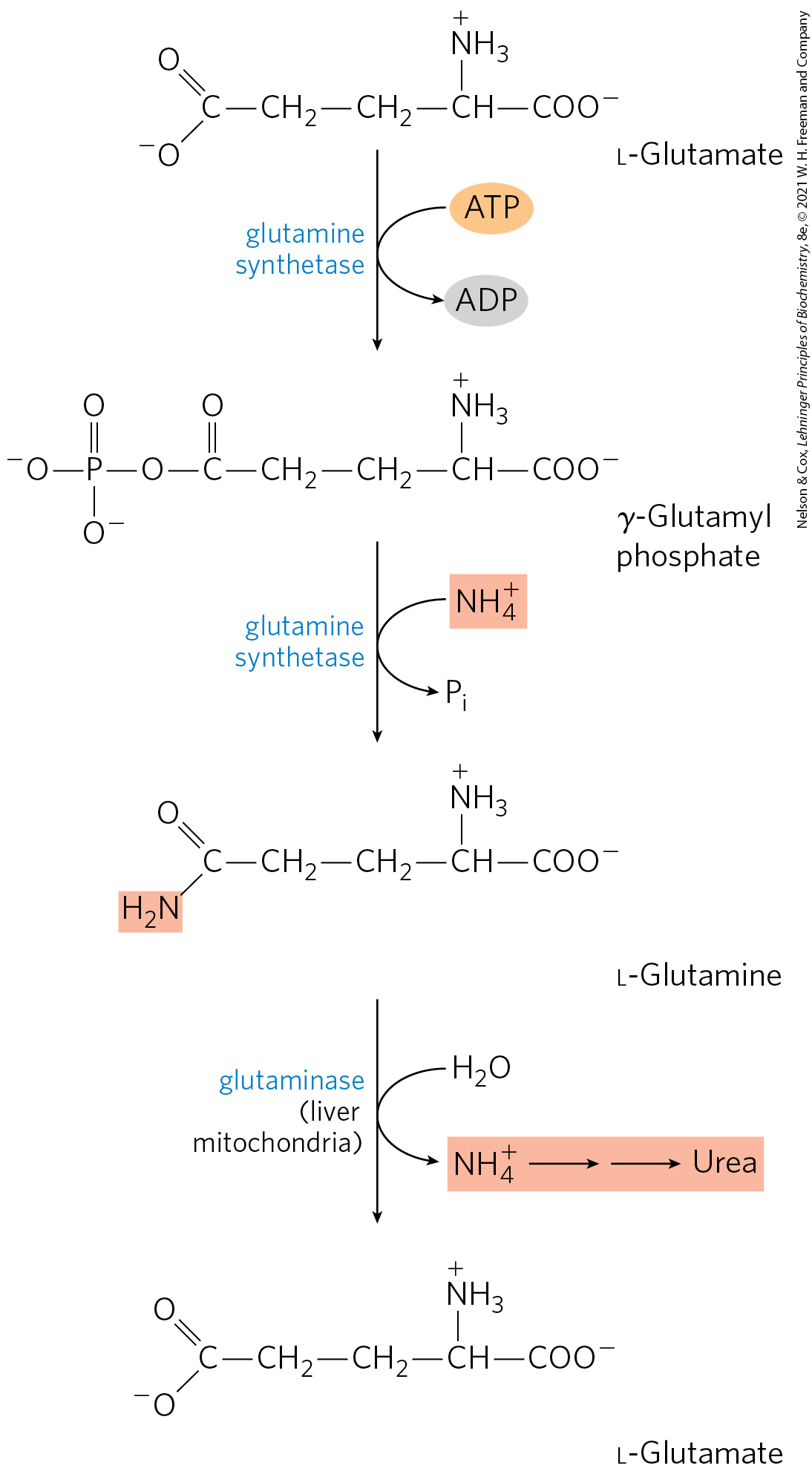
 Humans derive a small fraction of their oxidative energy from the catabolism of amino acids. Proteases degrade ingested proteins in the stomach and small intestine. Most proteases are initially synthesized as inactive zymogens.
Humans derive a small fraction of their oxidative energy from the catabolism of amino acids. Proteases degrade ingested proteins in the stomach and small intestine. Most proteases are initially synthesized as inactive zymogens.