4.3 Protein Tertiary and Quaternary Structures
The overall three-dimensional arrangement of all atoms in a protein is referred to as the protein’s tertiary structure. Whereas the term “secondary structure” refers to the spatial arrangement of amino acid residues that are adjacent in a segment of a polypeptide, tertiary structure includes longer-range aspects of amino acid sequence. Amino acids that are far apart in the polypeptide sequence and are in different types of secondary structure may interact within the completely folded structure of a protein. Interacting segments of polypeptide chains are held in their characteristic tertiary positions by several kinds of weak interactions (and sometimes by covalent bonds such as disulfide cross-links) between the segments. Some proteins contain two or more separate polypeptide chains, or subunits, which may be identical or different. The arrangement of these protein subunits in three-dimensional complexes constitutes quaternary structure.
 The overall three-dimensional arrangement of all atoms in a protein is referred to as the protein’s
The overall three-dimensional arrangement of all atoms in a protein is referred to as the protein’s In considering these higher levels of structure, it is useful to designate the major groups into which many proteins can be classified: fibrous proteins, with polypeptide chains arranged in long strands or sheets; globular proteins, with polypeptide chains folded into a spherical or globular shape; membrane proteins, with polypeptide chains embedded in hydrophobic lipid membranes; and intrinsically disordered proteins, with polypeptide chains lacking stable tertiary structures. We focus here on fibrous, globular, and intrinsically disordered proteins; membrane proteins are discussed in Chapter 11. These three groups are structurally distinct. Fibrous proteins usually consist of a single type of secondary structure, and their tertiary structure is relatively simple. Globular proteins often contain several types of secondary structure. Intrinsically disordered proteins can lack secondary structure entirely. The groups also differ functionally: the structures that provide support, shape, and external protection to vertebrates are made of fibrous proteins. Most enzymes are globular proteins, whereas regulatory proteins can be globular, disordered, or contain both globular and disordered segments.
Fibrous Proteins Are Adapted for a Structural Function
α-Keratin, collagen, and silk fibroin nicely illustrate the relationship between protein structure and biological function (Table 4-2). Fibrous proteins share properties that give strength and/or flexibility to the structures in which they occur. In each case, the fundamental structural unit is a simple repeating element of secondary structure. All fibrous proteins are insoluble in water, a property conferred by a high concentration of hydrophobic amino acid residues both in the interior of the protein and on its surface. These hydrophobic surfaces are largely buried, as many similar polypeptide chains are packed together to form elaborate supramolecular complexes. The underlying structural simplicity of fibrous proteins makes them particularly useful for illustrating some of the fundamental principles of protein structure discussed previously.
 Fibrous proteins share properties that give strength and/or flexibility to the structures in which they occur. In each case, the fundamental structural unit is a simple repeating element of secondary structure. All fibrous proteins are insoluble in water, a property conferred by a high concentration of hydrophobic amino acid residues both in the interior of the protein and on its surface. These hydrophobic surfaces are largely buried, as many similar polypeptide chains are packed together to form elaborate supramolecular complexes. The underlying structural simplicity of fibrous proteins makes them particularly useful for illustrating some of the fundamental principles of protein structure discussed previously.
Fibrous proteins share properties that give strength and/or flexibility to the structures in which they occur. In each case, the fundamental structural unit is a simple repeating element of secondary structure. All fibrous proteins are insoluble in water, a property conferred by a high concentration of hydrophobic amino acid residues both in the interior of the protein and on its surface. These hydrophobic surfaces are largely buried, as many similar polypeptide chains are packed together to form elaborate supramolecular complexes. The underlying structural simplicity of fibrous proteins makes them particularly useful for illustrating some of the fundamental principles of protein structure discussed previously.| Structure | Characteristics | Examples of occurrence |
|---|---|---|
α Helix, cross-linked by disulfide bonds |
Tough, insoluble protective structures of varying hardness and flexibility |
α-Keratin of hair, feathers, nails |
β Conformation |
Soft, flexible filaments |
Silk fibroin |
Collagen triple helix |
High tensile strength, without stretch |
Collagen of tendons, bone matrix |
α-Keratin The α-keratins have evolved for strength. Found only in mammals, these proteins constitute almost the entire dry weight of hair, wool, nails, claws, quills, horns, and hooves and much of the outer layer of skin. The α-keratins are part of a broader family of proteins called intermediate filament (IF) proteins. Other IF proteins are found in the cytoskeletons of animal cells. All IF proteins have a structural function and share the structural features exemplified by the α-keratins.
The α-keratin helix is a right-handed α helix, the same helix found in many other proteins. Francis Crick and Linus Pauling, in the early 1950s, independently suggested that the α helices of keratin were arranged as a coiled coil. Two strands of α-keratin, oriented in parallel (with their amino termini at the same end), are wrapped about each other to form a supertwisted coiled coil. The supertwisting amplifies the strength of the overall structure, just as strands are twisted to make a strong rope (Fig. 4-10). The twisting of the axis of an α helix to form a coiled coil explains the discrepancy between the 5.4 Å per turn predicted for an α helix by Pauling and Corey and the 5.15 to 5.2 Å repeating structure observed in the x-ray diffraction of hair (see end-of-chapter problem 2). The helical path of the supertwists is left-handed, opposite in sense to the α helix. The surfaces where the two α helices touch are made up of hydrophobic amino acid residues, their R groups meshed together in a regular interlocking pattern. This permits a close packing of the polypeptide chains within the left-handed supertwist. Not surprisingly, α-keratin is rich in the hydrophobic residues Ala, Val, Leu, Ile, Met, and Phe.
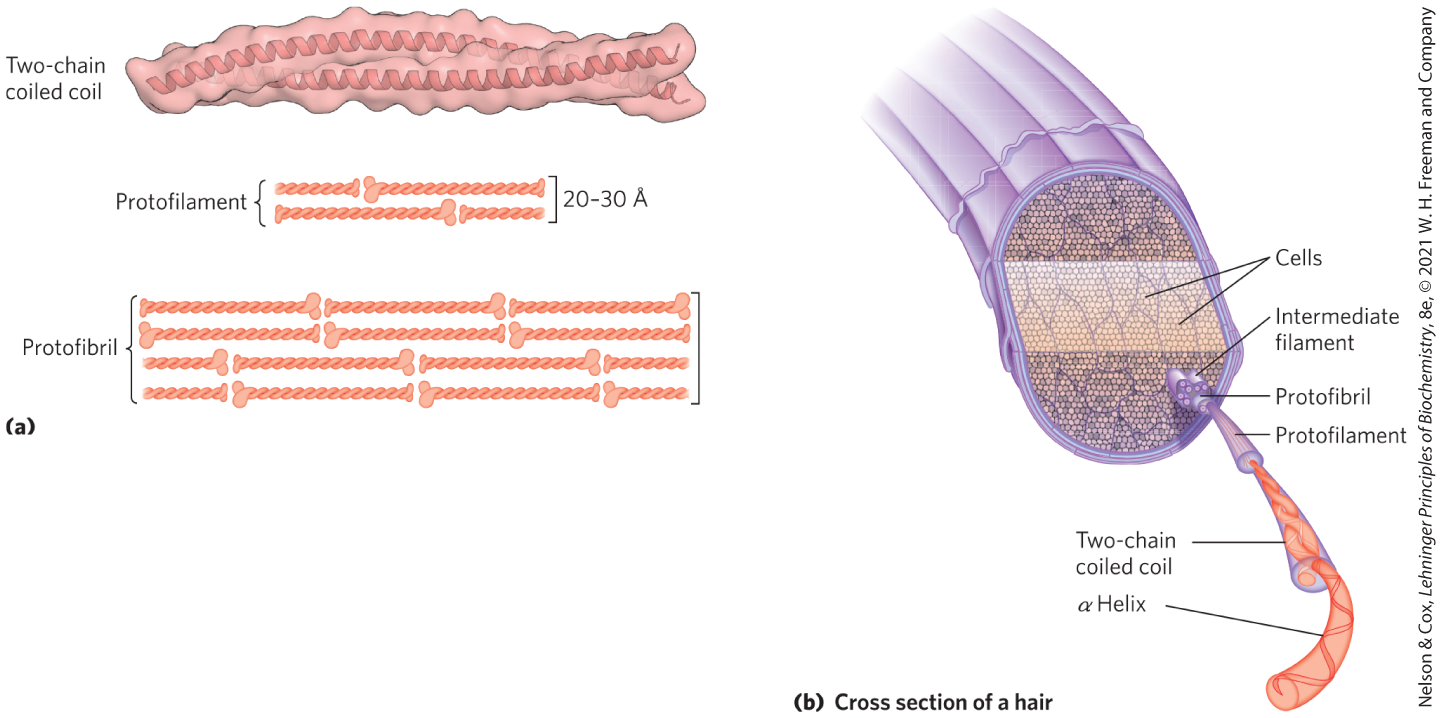
FIGURE 4-10 Structure of hair. (a) Hair α-keratin is an elongated α helix with somewhat thicker elements near the amino and carboxyl termini. Pairs of these helices are interwound in a left-handed sense to form two-chain coiled coils. These then combine in higher-order structures called protofilaments and protofibrils. About four protofibrils—32 strands of α-keratin in all—combine to form an intermediate filament. The individual two-chain coiled coils in the various substructures also seem to be interwound, but the handedness of the interwinding and other structural details are unknown. (b) A hair is an array of many α-keratin filaments, made up of the substructures shown in (a). [(a) Information from PDB ID 3TNU, C. H. Lee et al., Nature Struct. Mol. Biol. 19:707, 2012.]
An individual polypeptide in the α-keratin coiled coil has a relatively simple tertiary structure, dominated by an α-helical secondary structure with its helical axis twisted in a left-handed superhelix. The intertwining of the two α-helical polypeptides is an example of quaternary structure. Coiled coils of this type are common structural elements in filamentous proteins and in the muscle protein myosin (see Fig. 5-26). The quaternary structure of α-keratin can be quite complex. Many coiled coils can be assembled into large supramolecular complexes, such as the arrangement of α-keratin that forms the intermediate filament of hair (Fig. 4-10b).
The strength of fibrous proteins is enhanced by covalent cross-links between polypeptide chains in the multihelical “ropes” and between adjacent chains in a supramolecular assembly. In α-keratins, the cross-links stabilizing quaternary structure are disulfide bonds. In the hardest and toughest α-keratins, such as those of rhinoceros horn, up to 18% of the residues are cysteines involved in disulfide bonds.
Collagen Like the α-keratins, collagen has evolved to provide strength. It is found in connective tissue such as tendons, cartilage, the organic matrix of bone, and the cornea of the eye. In fact, collagen is the most abundant protein in mammals, usually comprising 25% to 35% of total protein content. The collagen helix is a unique secondary structure, quite distinct from the α helix. It is left-handed and has three amino acid residues per turn (Fig. 4-11 and Table 4-1). Collagen is also a coiled coil, but one with distinct tertiary and quaternary structures: three separate polypeptides, called α chains (not to be confused with α helices), are twisted about each other. The superhelical twisting is right-handed in collagen, opposite in sense to the left-handed helix of the α chains.
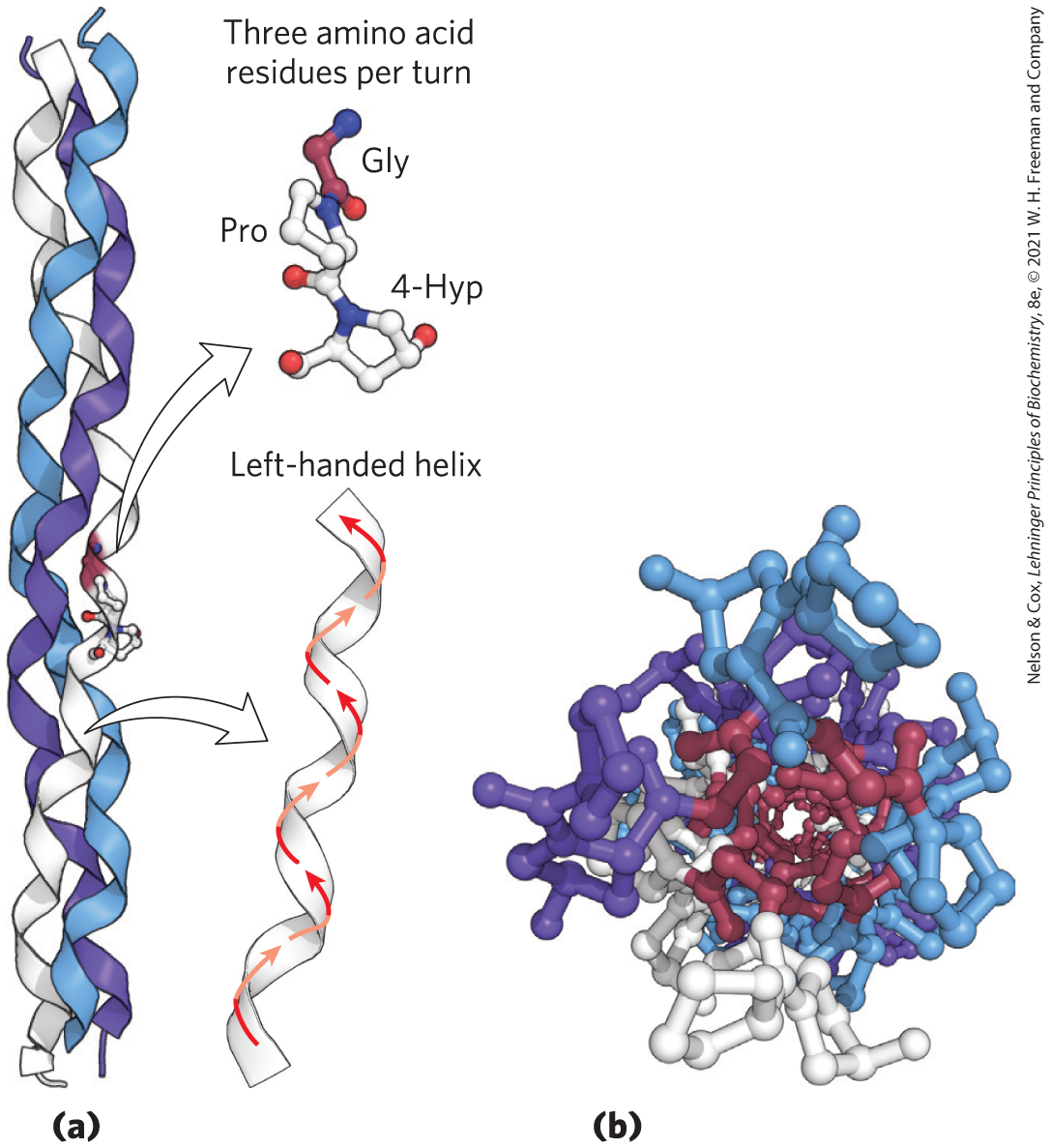
FIGURE 4-11 Structure of collagen. (a) The α chain of collagen has a repeating secondary structure unique to this protein. The repeating tripeptide sequence Gly–X–Y, where X is often Pro and Y is often 4-Hyp, adopts a left-handed helical structure with three residues per turn. Three of these helices (shown here in white, blue, and purple) wrap around one another with a right-handed twist. (b) The three-stranded collagen superhelix shown from one end, in a ball-and-stick representation. Gly residues are shown in red. Glycine, because of its small size, is required at the tight junction where the three chains are in contact. The balls in this illustration do not represent the van der Waals radii of the individual atoms. The center of the three-stranded superhelix is not hollow, as it appears here, but very tightly packed. [Data from PDB ID 1CGD, J. Bella et al., Structure 3:893, 1995.]
There are many types of vertebrate collagen. Typically, they contain about 35% Gly, 11% Ala, and 21% Pro and 4-Hyp (4-hydroxyproline, an uncommon amino acid; see Fig. 3-8a). The food product gelatin is derived from collagen. It has little nutritional value as a protein, because collagen is extremely low in many amino acids that are essential in the human diet. The unusual amino acid content of collagen is related to structural constraints unique to the collagen helix. The amino acid sequence in collagen is generally a repeating tripeptide unit, Gly–X–Y, where X is often Pro and Y is often 4-Hyp. Only Gly residues can be accommodated at the very tight junctions between the individual α chains (Fig. 4-11b). The Pro and 4-Hyp residues permit the sharp twisting of the collagen helix. The amino acid sequence and the supertwisted quaternary structure of collagen allow a very close packing of its three polypeptides. 4-Hydroxyproline has a special role in the structure of collagen — and in human history (Box 4-2).
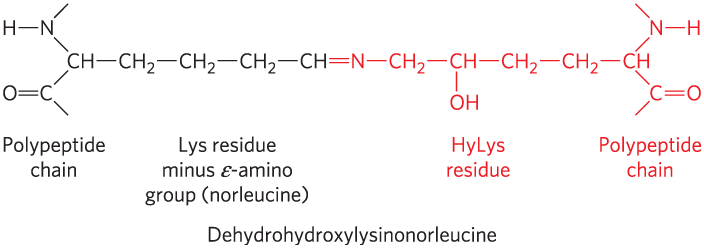
The tight wrapping of the α chains in the collagen triple helix provides tensile strength greater than that of a steel wire of equal cross section. Collagen fibrils (Fig. 4-12) are supramolecular assemblies consisting of triple-helical collagen molecules (sometimes referred to as tropocollagen molecules) associated in a variety of ways to provide different degrees of tensile strength. The α chains of collagen molecules and the collagen molecules of fibrils are cross-linked by unusual types of covalent bonds involving Lys, HyLys (5-hydroxylysine), or His residues that are present at a few of the X and Y positions. These links create uncommon amino acid residues such as dehydrohydroxylysinonorleucine. The increasingly rigid and brittle character of aging connective tissue results from accumulated covalent cross-links in collagen fibrils.
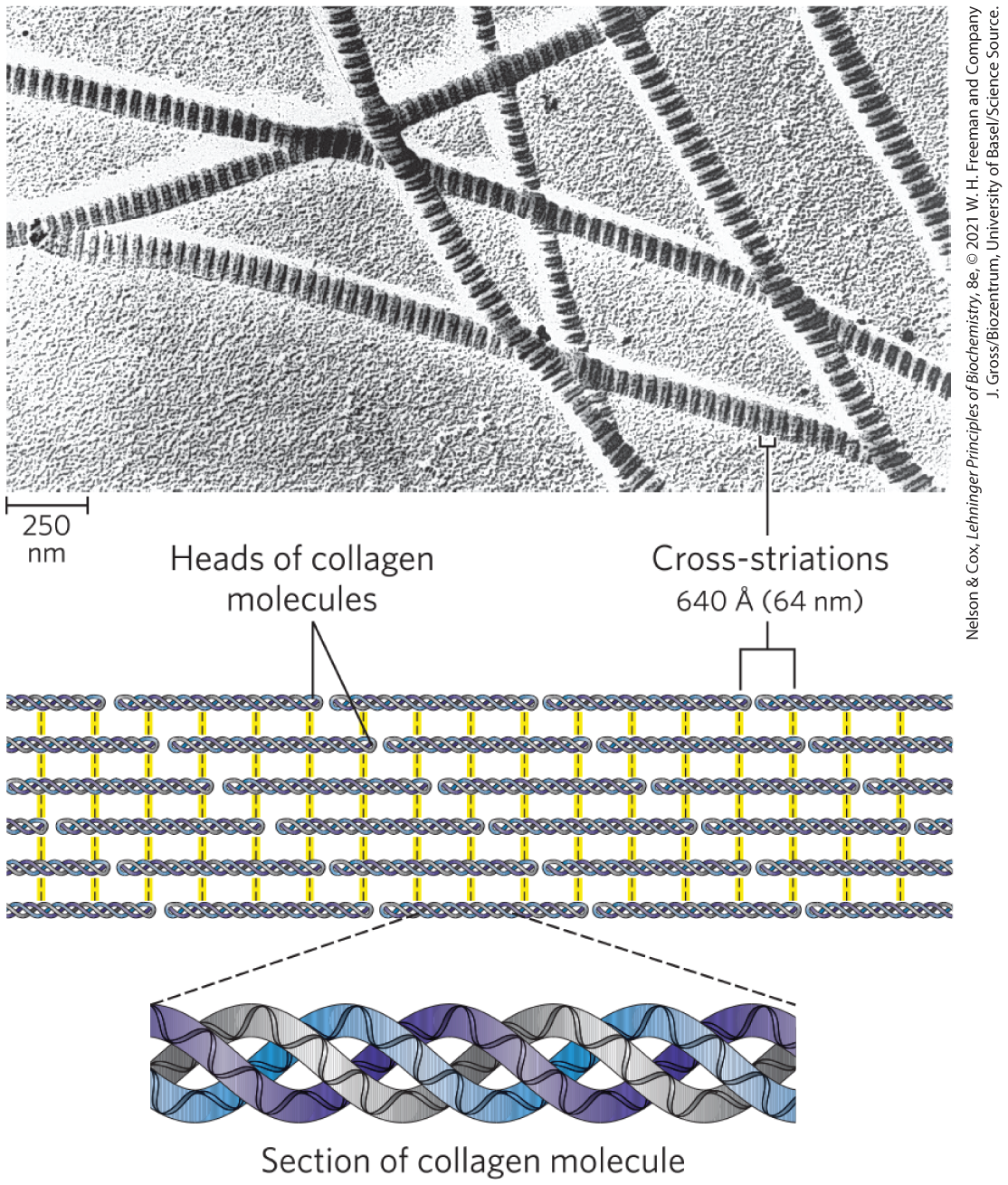
FIGURE 4-12 Structure of collagen fibrils. Collagen is a rod-shaped molecule, about 3,000 Å long and only 15 Å thick. Its three helically intertwined α chains may have different sequences; each chain has about 1,000 amino acid residues. Collagen fibrils are made up of collagen molecules aligned in a staggered fashion and cross-linked for strength. The specific alignment and degree of cross-linking vary with the tissue and produce characteristic cross-striations in an electron micrograph. In the example shown here, alignment of the head groups of every fourth molecule produces striations 640 Å(64 nm) apart.
A typical mammal has more than 30 structural variants of collagen, particular to certain tissues and each somewhat different in sequence and function. Some human genetic defects in collagen structure illustrate the close relationship between amino acid sequence and three-dimensional structure in this protein. Osteogenesis imperfecta is characterized by abnormal bone formation in babies; at least eight variants of this condition, with different degrees of severity, occur in the human population. Ehlers-Danlos syndrome is characterized by loose joints, and at least six variants occur in humans. The composer Niccolò Paganini (1782–1840) was famed for his seemingly impossible dexterity in playing the violin. He suffered from a variant of Ehlers-Danlos syndrome that rendered him effectively double-jointed. In both disorders, some variants can be lethal, whereas others cause lifelong problems.
 A typical mammal has more than 30 structural variants of collagen, particular to certain tissues and each somewhat different in sequence and function. Some human genetic defects in collagen structure illustrate the close relationship between amino acid sequence and three-dimensional structure in this protein. Osteogenesis imperfecta is characterized by abnormal bone formation in babies; at least eight variants of this condition, with different degrees of severity, occur in the human population. Ehlers-Danlos syndrome is characterized by loose joints, and at least six variants occur in humans. The composer Niccolò Paganini (1782–1840) was famed for his seemingly impossible dexterity in playing the violin. He suffered from a variant of Ehlers-Danlos syndrome that rendered him effectively double-jointed. In both disorders, some variants can be lethal, whereas others cause lifelong problems.
A typical mammal has more than 30 structural variants of collagen, particular to certain tissues and each somewhat different in sequence and function. Some human genetic defects in collagen structure illustrate the close relationship between amino acid sequence and three-dimensional structure in this protein. Osteogenesis imperfecta is characterized by abnormal bone formation in babies; at least eight variants of this condition, with different degrees of severity, occur in the human population. Ehlers-Danlos syndrome is characterized by loose joints, and at least six variants occur in humans. The composer Niccolò Paganini (1782–1840) was famed for his seemingly impossible dexterity in playing the violin. He suffered from a variant of Ehlers-Danlos syndrome that rendered him effectively double-jointed. In both disorders, some variants can be lethal, whereas others cause lifelong problems.All of the variants of both conditions result from the substitution of an amino acid residue with a larger R group (such as Cys or Ser) for a single Gly residue in an α chain in one or another of the collagen proteins (a different Gly residue in each disorder). These single-residue substitutions have a catastrophic effect on collagen function because they disrupt the Gly–X–Y repeat that gives collagen its unique helical structure. Given its role in the collagen triple helix (Fig. 4-11), Gly cannot be replaced by another amino acid residue without substantial deleterious effects on collagen structure.
Fibroin The protein of silk, fibroin, is produced by insects and spiders. Its polypeptide chains are predominantly in the β conformation. Fibroin is rich in Ala and Gly residues, permitting a close packing of β sheets and an interlocking arrangement of R groups (Fig. 4-13). The overall structure is stabilized by extensive hydrogen bonding between all peptide linkages in the polypeptides of each β sheet and by the optimization of van der Waals interactions between sheets. Silk does not stretch, because the β conformation is already highly extended (Fig. 4-5). However, the structure is flexible, because the sheets are held together by numerous weak interactions rather than by covalent bonds such as the disulfide bonds in α-keratins.
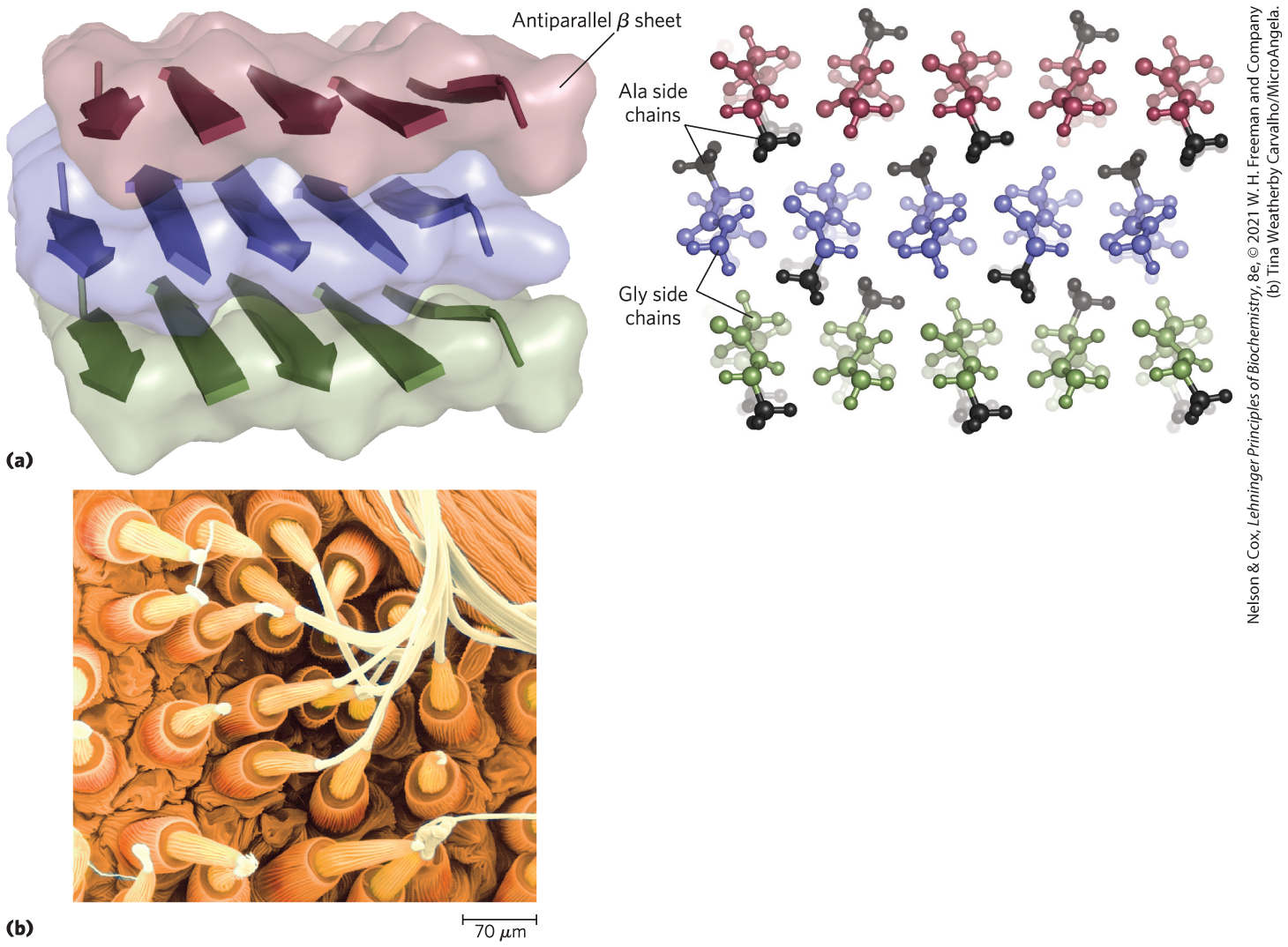
FIGURE 4-13 Structure of silk. The fibers in silk cloth and in a spider web are made up primarily of the protein fibroin. (a) Fibroin consists of layers of antiparallel β sheets rich in Ala and Gly residues. The small side chains interdigitate and allow close packing of the sheets, as shown in the ball-and-stick view. The segments shown here would be just a small part of the fibroin strand. (b) Strands of silk emerge from the spinnerets of a spider in this colorized scanning electron micrograph. [(a) Data from PDB ID 1SLK, S. A. Fossey et al., Biopolymers 31:1529, 1991. (b) Tina Weatherby Carvalho/MicroAngela.]
Structural Diversity Reflects Functional Diversity in Globular Proteins
In a globular protein, different segments of the polypeptide chain (or multiple polypeptide chains) fold back on each other, generating a more compact shape than is seen in the fibrous proteins (Fig. 4-14). The folding also provides the structural diversity necessary for proteins to carry out a wide array of biological functions. Globular proteins include enzymes, transport proteins, motor proteins, regulatory proteins, immunoglobulins, and proteins with many other functions.

FIGURE 4-14 Globular protein structures are compact and varied. Human serum albumin has 585 residues in a single chain. Given here are the approximate dimensions its single polypeptide chain would have if it occurred entirely in extended β conformation or as an α helix. Also shown is the size of the protein in its native globular form, as determined by x-ray crystallography; the polypeptide chain must be very compactly folded to fit into these dimensions.
Our discussion of globular proteins begins with the principles gleaned from the first protein structures to be elucidated. This is followed by a detailed description of protein substructure and comparative categorization. Such discussions are possible only because of the vast amount of information available online from publicly accessible databases, particularly the Protein Data Bank, or PDB (Box 4-3).
Myoglobin Provided Early Clues about the Complexity of Globular Protein Structure
The first breakthrough in understanding the three-dimensional structure of a globular protein came from x-ray diffraction studies of myoglobin carried out by John Kendrew and his colleagues in the 1950s. Myoglobin is a relatively small oxygen-binding protein of muscle cells. It functions both to store oxygen and to facilitate oxygen diffusion in rapidly contracting muscle tissue. Myoglobin contains a single polypeptide chain of 153 amino acid residues of known sequence and a single iron protoporphyrin, or heme, group. The same heme group that is found in myoglobin is found in hemoglobin, the oxygen-binding protein of erythrocytes, and is responsible for the deep red-brown color of both myoglobin and hemoglobin. Myoglobin is particularly abundant in the muscles of diving mammals such as whales, seals, and porpoises — so abundant that the muscles of these animals are brown. Storage and distribution of oxygen by muscle myoglobin permits diving mammals to remain submerged for long periods. The activities of myoglobin and other globin molecules are investigated in greater detail in Chapter 5.
Figure 4-15 shows several structural representations of myoglobin, illustrating how the polypeptide chain is folded in three dimensions — its tertiary structure. The red group surrounded by protein is heme. The backbone of the myoglobin molecule consists of eight relatively straight segments of α helix interrupted by bends, some of which are β turns. The longest α helix has 23 amino acid residues and the shortest has only 7; all helices are right-handed. More than 70% of the residues in myoglobin are in these α-helical regions. X-ray analysis has revealed the precise position of each of the R groups, which fill up nearly all the space within the folded chain that is not occupied by backbone atoms.
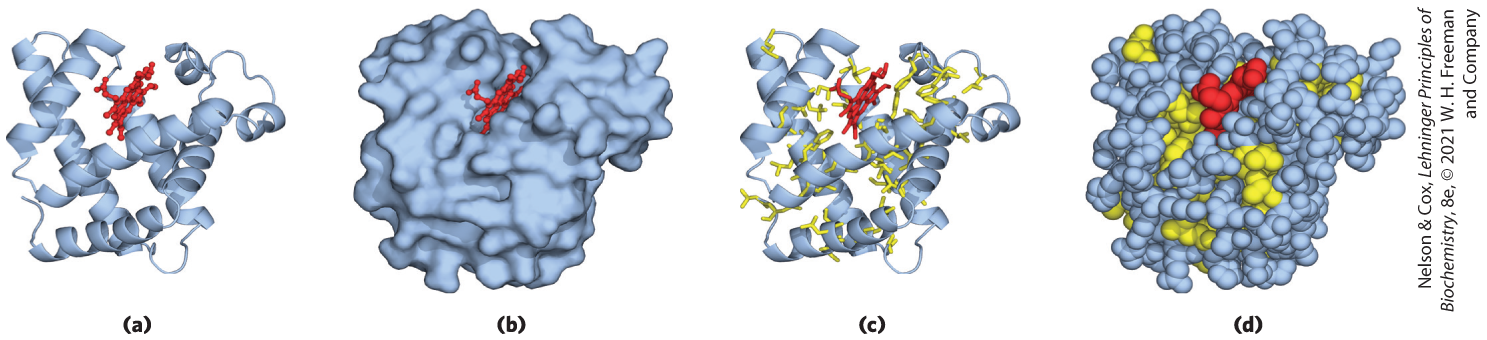
FIGURE 4-15 Tertiary structure of sperm whale myoglobin. Orientation of the protein is similar in (a) through (d); the heme group is shown in red. In addition to illustrating the myoglobin structure, this figure provides examples of several different ways to display protein structure. (a) The polypeptide backbone in a ribbon representation of a type introduced by Jane Richardson, which highlights regions of secondary structure. The α-helical regions are evident. (b) Surface contour image; this is useful for visualizing pockets in the protein where other molecules might bind. (c) Ribbon representation including side chains (yellow) for the hydrophobic residues Leu, Ile, Val, and Phe. (d) Space-filling model with all amino acid side chains. Each atom is represented by a sphere encompassing its van der Waals radius. The hydrophobic residues are again shown in yellow; most are buried in the interior of the protein and thus are not visible. [Data from PDB ID 1MBO, S. E. Phillips, J. Mol. Biol. 142:531, 1980.]
Many important conclusions were drawn from the structure of myoglobin. The positioning of amino acid side chains reflects a structure that is largely stabilized by the hydrophobic effect. Most of the hydrophobic R groups are in the interior of the molecule, hidden from exposure to water. All but two of the polar R groups are located on the outer surface of the molecule, and all are hydrated. The myoglobin molecule is so compact that its interior has room for only four molecules of water. This dense hydrophobic core is typical of globular proteins. The fraction of space occupied by atoms in an organic liquid is 0.4 to 0.6. In a globular protein the fraction is about 0.75, comparable to that in a crystal (in a typical crystal the fraction is 0.70 to 0.78, near the theoretical maximum). In this packed environment, weak interactions strengthen and reinforce each other. For example, the nonpolar side chains in the core are so close together that short-range van der Waals interactions make a significant contribution to stabilizing interactions.
Deduction of the structure of myoglobin confirmed some expectations and introduced some new elements of secondary structure. As predicted by Pauling and Corey, all the peptide bonds are in the planar trans configuration. The α helices in myoglobin provided the first direct experimental evidence for the existence of this type of secondary structure. Three of the four Pro residues are found at bends. The fourth Pro residue occurs within an α helix, where it creates a kink necessary for tight helix packing.
The flat heme group rests in a crevice, or pocket, in the myoglobin molecule. Within this pocket, the accessibility of the heme group to solvent is highly restricted. This is important for function, because free heme groups in an oxygenated solution are rapidly oxidized from the ferrous form, which is active in the reversible binding of , to the ferric form, which does not bind . As myoglobin structures from many different species were resolved, investigators were able to observe the structural changes that accompany the binding of oxygen or other molecules and thus, for the first time, to understand the correlation between protein structure and function. Hundreds of proteins have now been subjected to similar analysis.
Globular Proteins Have a Variety of Tertiary Structures
Myoglobin illustrates just one of many ways in which a polypeptide chain can fold. Table 4-3 shows the proportions of α helix and β conformation (expressed as percentage of residues in each type) in several small, single-chain, globular proteins. Each of these proteins has a distinct structure, adapted for its particular biological function, but together they share several important properties with myoglobin. Each is folded compactly, and in each case the hydrophobic amino acid side chains are oriented toward the interior (away from water) and the hydrophilic side chains are on the surface. The structures are also stabilized by a multitude of hydrogen bonds and some ionic interactions.
 Each is folded compactly, and in each case the hydrophobic amino acid side chains are oriented toward the interior (away from water) and the hydrophilic side chains are on the surface. The structures are also stabilized by a multitude of hydrogen bonds and some ionic interactions.
Each is folded compactly, and in each case the hydrophobic amino acid side chains are oriented toward the interior (away from water) and the hydrophilic side chains are on the surface. The structures are also stabilized by a multitude of hydrogen bonds and some ionic interactions.| Residues (%)a | ||
|---|---|---|
| Protein (total residues) | α Helix | β Conformation |
Chymotrypsin (247) |
14 |
45 |
Ribonuclease (124) |
26 |
35 |
Carboxypeptidase (307) |
38 |
17 |
Cytochrome c (104) |
39 |
0 |
Lysozyme (129) |
40 |
12 |
Myoglobin (153) |
78 |
0 |
|
Source: Data from C. R. Cantor and P. R. Schimmel, Biophysical Chemistry, Part I: The Conformation of Biological Macromolecules, p. 100, W. H. Freeman and Company, 1980. aPortions of the polypeptide chains not accounted for by α helix or β conformation consist of bends and irregularly coiled or extended stretches. Segments of α helix and β conformation sometimes deviate slightly from their normal dimensions and geometry. |
||
To understand a complete three-dimensional structure, we need to analyze its folding patterns. We begin by defining two important terms that describe protein structural patterns or elements in a polypeptide chain; then we turn to the folding rules. The first term is motif, also called a fold. A motif or fold is a recognizable folding pattern involving two or more elements of secondary structure and the connection(s) between them. A motif can be very simple, such as two elements of secondary structure folded against each other, and may represent only a small part of a protein. An example is a β-α-β loop (Fig. 4-16a). A motif can also be a very elaborate structure involving scores of protein segments folded together, such as the β barrel (Fig. 4-16b). In some cases, a single large motif may comprise the entire protein. The terms “motif” and “fold” are often used interchangeably, although “fold” is applied more commonly to somewhat more complex folding patterns. The segment defined as a motif or a fold may or may not be independently stable. We have already encountered a well-studied motif, the coiled coil of α-keratin, which is also found in some other proteins. The distinctive arrangement of eight α helices in myoglobin is replicated in all globins and is called the globin fold. Note that a motif is not a hierarchical structural element falling between secondary and tertiary structure. It is simply a folding pattern.
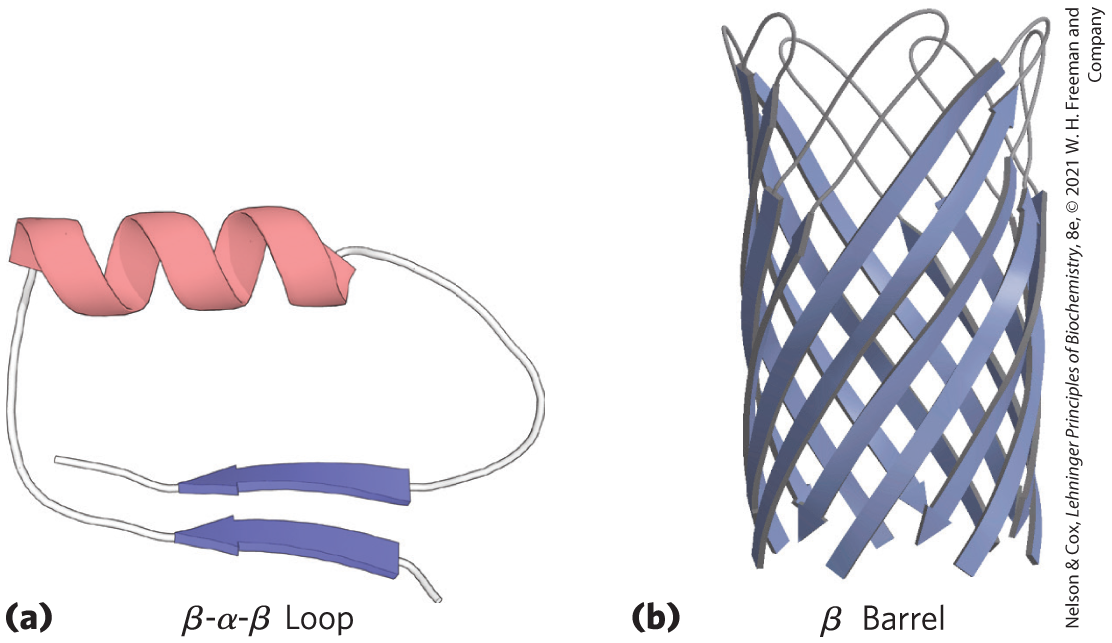
FIGURE 4-16 Motifs. (a) A simple motif, the β-α-β loop. (b) A more elaborate motif, the β barrel. This β barrel is a single domain of α-hemolysin (a toxin that kills a cell by creating a hole in its membrane) from the bacterium Staphylococcus aureus. [Data from (a) PDB ID 4TIM, M. E. Noble et al., J. Med. Chem., 34:2709, 1991; (b) PDB ID 7AHL, L. Song et al., Science 274:1859, 1996.]
The second term for describing structural patterns is domain. A domain, as defined by Jane Richardson in 1981, is a part of a polypeptide chain that is independently stable or could undergo movements as a single entity with respect to the entire protein. Polypeptides with more than a few hundred amino acid residues often fold into two or more domains, sometimes with different functions. In many cases, a domain from a large protein will retain its native three-dimensional structure even when separated (for example, by proteolytic cleavage) from the remainder of the polypeptide chain. In a protein with multiple domains, each domain may appear as a distinct globular lobe (Fig. 4-17); more commonly, extensive contacts between domains make individual domains hard to discern. Different domains often have distinct functions, such as the binding of small molecules or interaction with other proteins. Small proteins usually have only one domain (the domain is the protein).
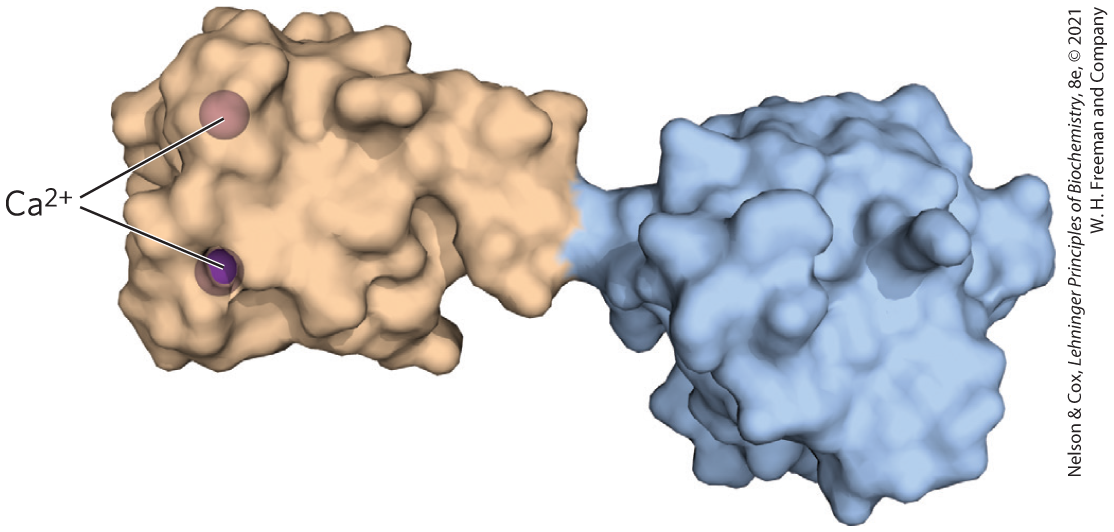
FIGURE 4-17 Structural domains in the polypeptide troponin C. This calcium-binding protein, associated with muscle, has two separate calcium-binding domains, shown here in brown and blue. [Data from PDB ID 4TNC, K. A. Satyshur et al., J. Biol. Chem. 263:1628, 1988.]
Folding of polypeptides is subject to an array of physical and chemical constraints, and several rules have emerged from studies of common protein-folding patterns.
- The hydrophobic effect makes a large contribution to the stability of protein structures. Burial of hydrophobic amino acid R groups so as to exclude water requires at least two layers of secondary structure. Simple motifs such as the β-α-β loop (Fig. 4-16a) create two such layers.
- Where they occur together in a protein, α helices and β sheets generally are found in different structural layers. This is because the backbone of a polypeptide segment in the β conformation (Fig. 4-5) cannot readily hydrogen-bond to an α helix that is adjacent to it.
- Segments adjacent to each other in the amino acid sequence are usually stacked adjacent to each other in the folded structure. Distant segments of a polypeptide may come together in the tertiary structure, but this is not the norm.
- The β conformation is most stable when the individual segments are twisted slightly in a right-handed sense. This influences both the arrangement of β sheets derived from the twisted segments and the path of the polypeptide connections between them. Two parallel β strands, for example, must be connected by a crossover strand (Fig. 4-18a). In principle, this crossover could have a right-handed or left-handed conformation, but in proteins it is almost always right-handed. Right-handed connections tend to be shorter than left-handed connections and tend to bend through smaller angles, making them easier to form. The twisting of β sheets also leads to a characteristic twisting of the structure formed by many such segments together, as seen in the β barrel (Fig. 4-16b) and the twisted β sheet (Fig. 4-18c), which form the core of many larger structures.
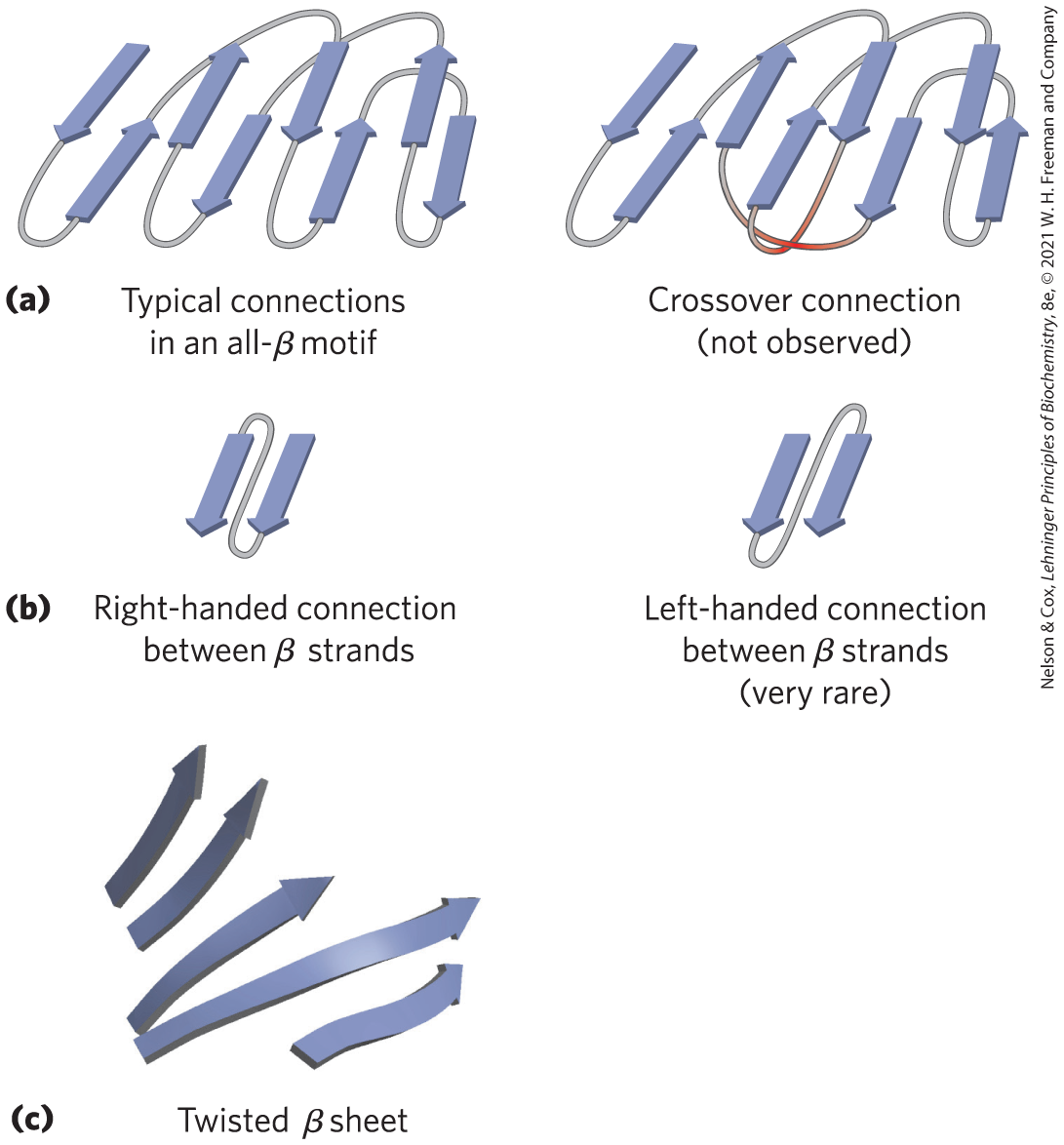
FIGURE 4-18 Stable folding patterns in proteins. (a) Connections between β strands in layered β sheets. The strands here are viewed from one end, with no twisting. The connections at a given end (e.g., near the viewer) rarely cross one another. An example of such a rare crossover is illustrated by the red strands in the structure on the right. (b) Because of the right-handed twist in β strands, connections between strands are generally right-handed. Left-handed connections must traverse sharper angles and are harder to form. (c) This twisted β sheet is from a domain of photolyase (a protein that repairs certain types of DNA damage) from E. coli. Connecting loops have been removed so as to focus on the folding of the β sheet. [Data from PDB ID 1DNP, H. W. Park et al., Science 268:1866, 1995.]
Following these rules, complex motifs can be built up from simple ones. For example, a series of β-α-β loops arranged so that the β strands form a barrel creates a particularly stable and common motif, the α/β barrel (Fig. 4-19). In this structure, each parallel β segment is attached to its neighbor by an α-helical segment. All connections are right-handed. The α/β barrel is found in many enzymes, often with a binding site (for a cofactor or a substrate) in the form of a pocket near one end of the barrel. Note that domains with similar folding patterns are said to have the same motif, even though their constituent α helices and β sheets may differ in length.
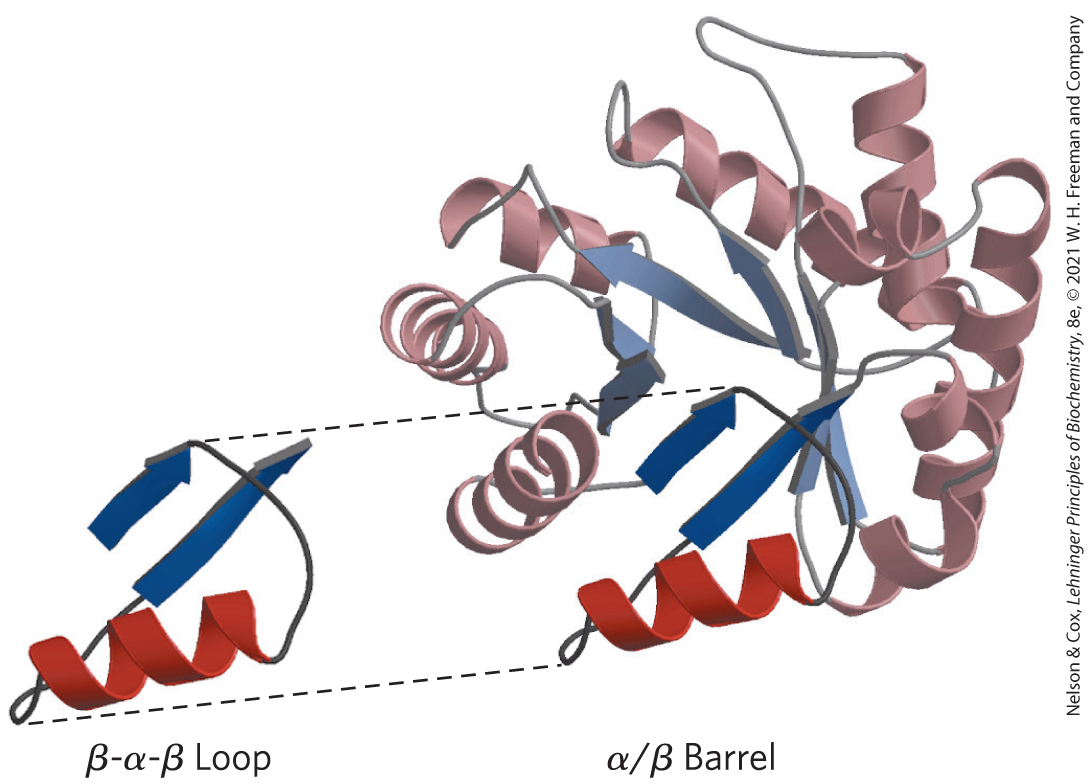
FIGURE 4-19 Constructing large motifs from smaller ones. The α/β barrel is a commonly occurring motif constructed from repetitions of the β-α-β loop motif. This α/β barrel is a domain of pyruvate kinase (a glycolytic enzyme) from rabbit. [Data from PDB ID 1PKN, T. M. Larsen et al., Biochemistry 33:6301, 1994.]
Some Proteins or Protein Segments Are Intrinsically Disordered
Although many proteins contain well-folded and stable structures, this is not necessary for the biological function of all proteins. Many proteins or protein segments lack ordered structures in solution. The concept that some proteins function in the absence of a definable three-dimensional structure comes from reassessment of data from many different proteins. As many as a third of all human proteins may be unstructured or may have significant unstructured segments. All organisms have some proteins that fall into this category. Intrinsically disordered proteins have properties that are distinct from those of classical, structured proteins. They often lack a hydrophobic core and instead are characterized by high densities of charged amino acid residues such as Lys, Arg, and Glu. Pro residues are also prominent, as they tend to disrupt ordered structures.
Structural disorder and high charge density can facilitate the function of some proteins as spacers, insulators, or linkers in larger structures. Other disordered proteins are scavengers, binding up ions and small molecules in solution and serving as reservoirs or garbage dumps. However, many intrinsically disordered proteins are at the heart of important protein interaction networks. The lack of an ordered structure can facilitate a kind of functional promiscuity, allowing one protein to interact with multiple or even dozens of partners. Structural disorder allows some inhibitor proteins, such as the mammalian cell division protein p27, to interact with multiple targets in different ways. In solution, p27 lacks definable structure. However, it wraps around and inhibits the action of several enzymes called protein kinases (see Chapter 6) that facilitate cell division. The flexible structure of p27 allows it to accommodate itself to its different target proteins. Human tumor cells, which are cells that have lost the capacity to control cell division normally, generally have reduced levels of p27; the lower the levels of p27, the poorer the prognosis for the cancer patient.
Similarly, intrinsically disordered proteins are often present as hubs or scaffolds at the center of protein networks that constitute signaling pathways (see Fig. 12-30). These proteins, or parts of them, may interact with many different binding partners. They often take on an ordered structure when they interact with other proteins, but the structure they assume may vary with different binding partners. The mammalian protein p53 is also critical in the control of cell division. It contains both structured and unstructured segments, and the different segments interact with dozens of other proteins. An unstructured region of p53 at the carboxyl terminus interacts with at least four different binding partners and assumes a different structure in each of the complexes (Fig. 4-20).
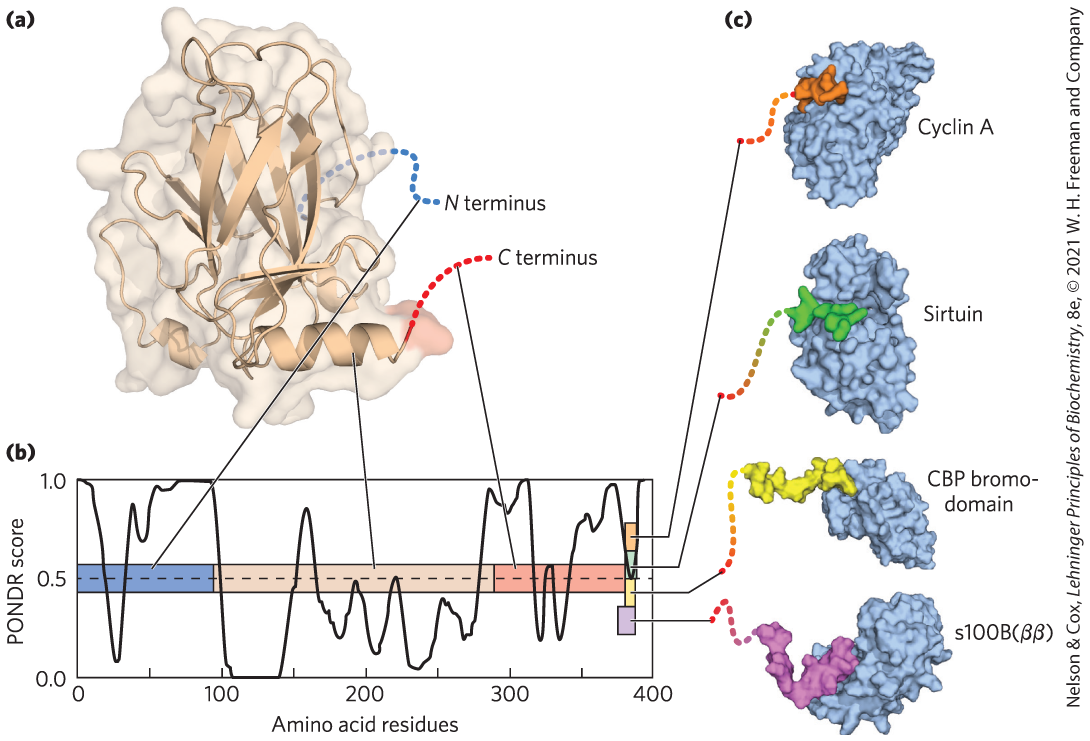
FIGURE 4-20 Binding of the intrinsically disordered carboxyl terminus of p53 protein to its binding partners. (a) The p53 protein is made up of several different segments. Only the central domain is well ordered. (b) The linear sequence of the p53 protein is depicted as a colored bar. The overlaid graph presents a plot of the PONDR (Predictor of Natural Disordered Regions) score versus the protein sequence. PONDR is one of the best available algorithms for predicting the likelihood that a given amino acid residue is in a region of intrinsic disorder, based on the surrounding amino acid sequence and amino acid composition. A score of 1.0 indicates a probability of 100% that a protein will be disordered. In the actual protein structure, the tan central domain is ordered. The amino-terminal (blue) and carboxyl-terminal (red) regions are disordered. (c) The very end of the carboxyl-terminal region has multiple binding partners and folds when it binds to each of them; however, the three-dimensional structure that is assumed when binding occurs is different for each of the interactions shown, and thus this carboxyl-terminal segment (11 to 20 residues) is shown in a different color in each complex. [Information from V. N. Uversky, Intl. J. Biochem. Cell Biol. 43:1090, 2011, Fig. 5. (a) Data from PDB ID 1TUP, Y. Cho et al., Science 265:346, 1994. (c) Data from Cyclin A: PDB ID 1H26, E. D. Lowe et al., Biochemistry 41:15,625, 2002; sirtuin: PDB ID 1MA3, J. L. Avalos et al., Mol. Cell 10:523, 2002; CBP bromodomain: PDB ID 1JSP, S. Mujtaba et al., Mol. Cell 13:251, 2004; s100B(ββ): PDB ID 1DT7, R. R. Rustandi et al., Nature Struct. Biol. 7:570, 2000.]
Protein Motifs Are the Basis for Protein Structural Classification
More than 150,000 structures are now archived in the Protein Data Bank (PDB; for a deeper explanation, see Box 4-3). An enormous amount of information about protein structural principles, protein function, and protein evolution is contained in these data. Other databases have organized this information and made it more readily accessible. In the Structural Classification of Proteins database, or SCOP2 (http://scop2.mrc-lmb.cam.ac.uk), all of the protein information in the PDB can be searched within four different categories: (1) protein relationships, (2) structural classes, (3) protein types, and (4) evolutionary events. Figure 4-21 presents examples of protein motifs taken from SCOP2 to illustrate the potential of searching within each category. The figure also introduces another way to represent elements of secondary structure and the relationships among segments of secondary structure in a protein — the topology diagram.
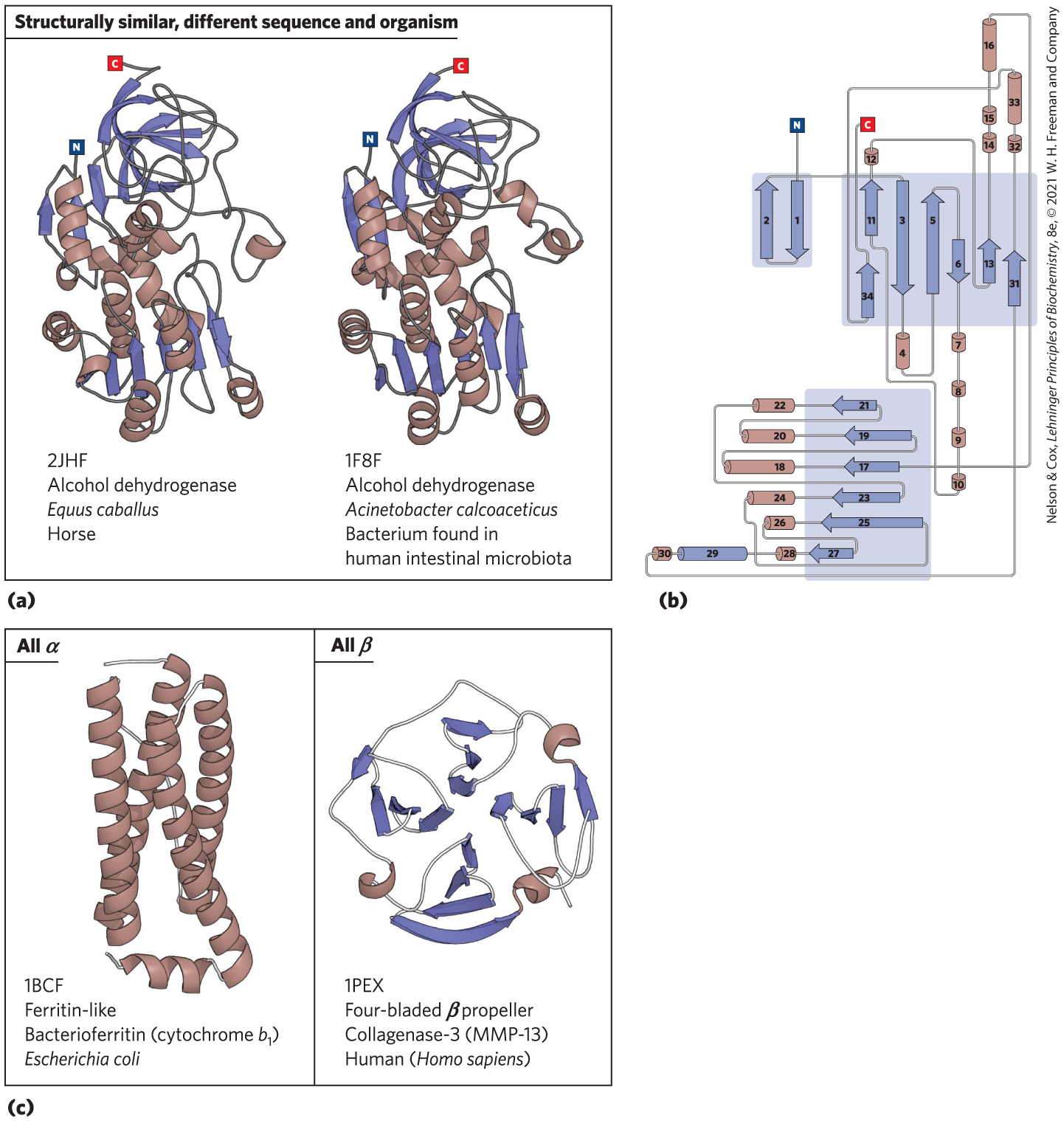
FIGURE 4-21 Organization of proteins based on motifs. A few of the hundreds of known stable motifs. (a) Structural diagrams of the enzyme alcohol dehydrogenase from two different organisms. Such comparisons illustrate evolutionary relationships that conserve structure as well as function. (b) A topology diagram for the alcohol dehydrogenase from Acinetobacter calcoaceticus. Topology diagrams provide a way to visualize elements of secondary structure and their interconnections in two dimensions; the diagrams can be very useful in comparing structural folds or motifs. (c) The Structural Classification of Proteins (SCOP2) database (http://scop2.mrc-lmb.cam.ac.uk) organizes protein folds into four classes: all α, all β, α/β, and α + β. Examples of all α folds and all β folds are shown with their structural classification data (PDB ID, fold name, protein name, and source organism) from the SCOP2 database. The PDB ID is the unique accession code given to each structure archived in the Protein Data Bank (www.rcsb.org). [Data from (a) PDB ID 2JHF, R. Meijers et al., Biochemistry 46:5446, 2007; (a, b) PDB ID 1F8F, J. C. Beauchamp et al. (c) PDB ID 1BCF, F. Frolow et al., Nature Struct. Biol. 1:453, 1994; PDB ID 1PEX, F. X. Gomis-Ruth et al., J. Mol. Biol. 264:556, 1996.]
The number of folding patterns is not infinite. Among the tens of thousands of distinct protein structures archived in the PDB, only about 1,400 different folds or motifs are classified by the SCOP2 database. Given the many years of progress in structural biology, new motifs are now discovered only rarely. Many examples of recurring domain or motif structures are available, and these reveal that protein tertiary structure is more reliably conserved than amino acid sequence. The comparison of protein structures can thus provide much information about evolution. Proteins with significant similarity in primary structure and/or with similar tertiary structure and function are said to be in the same protein family. The protein structures in the PDB belong to about 4,000 different protein families. A strong evolutionary relationship is usually evident within a protein family. For example, the globin family has many different proteins with both structural and sequence similarities to myoglobin (as seen in the proteins used as examples in Figures 4-30 and 4-31 and in Chapter 5). Two or more families that have little similarity in amino acid sequence but make use of the same major structural motif and have functional similarities are grouped into superfamilies. An evolutionary relationship among families in a superfamily is considered probable, even though time and functional distinctions — that is, different adaptive pressures — may have erased many of the telltale sequence relationships.
A protein family may be widespread in all three domains of cellular life — the Bacteria, Archaea, and Eukarya — suggesting an ancient origin. Many proteins involved in intermediary metabolism and the metabolism of nucleic acids and proteins fall into this category. Other families may be present in only a small group of organisms, indicating that the structure arose more recently. Tracing the natural history of structural motifs through the use of structural classifications in databases such as SCOP2 provides a powerful complement to sequence analyses in tracing evolutionary relationships. The SCOP2 database is curated manually, with the objective of placing proteins in the correct evolutionary framework based on conserved structural features.
Structural motifs become especially important in defining protein families and superfamilies. Improved protein classification and comparison systems lead inevitably to the elucidation of new functional relationships. Given the central role of proteins in living systems, these structural comparisons can help illuminate every aspect of biochemistry, from the evolution of individual proteins to the evolutionary history of complete metabolic pathways.
Protein Quaternary Structures Range from Simple Dimers to Large Complexes
Many proteins have multiple polypeptide subunits (from two to hundreds). The association of polypeptide chains can serve a variety of functions. Many multisubunit proteins have regulatory roles; the binding of small molecules may affect the interaction between subunits, causing large changes in the protein’s activity in response to small changes in the concentration of substrate or regulatory molecules (Chapter 6). In other cases, separate subunits take on separate but related functions, such as catalysis and regulation. Some associations, such as those seen in the fibrous proteins considered earlier in this chapter and the coat proteins of viruses, serve primarily structural roles. Some very large protein assemblies are the site of complex, multistep reactions. For example, each ribosome, the site of protein synthesis, incorporates dozens of protein subunits along with RNA molecules.
A multisubunit protein can also be referred to as an oligomer or multimer. If an oligomer has nonidentical subunits, the overall structure of the protein can be asymmetric and quite complicated. However, many oligomers have identical subunits or repeating groups of nonidentical subunits, usually in symmetric arrangements. As noted in Chapter 3, the repeating structural unit in such an oligomeric protein, whether a single subunit or a group of subunits, is called a protomer.
The first oligomeric protein to have its three-dimensional structure determined was hemoglobin which contains four polypeptide chains and four heme prosthetic groups, in which the iron atoms are in the ferrous state (as we shall see in Chapter 5). The protein portion, the globin, consists of two α chains (141 residues each) and two β chains (146 residues each). Note that in this case, α and β do not refer to secondary structures. In a practice that can be confusing to the beginning student, the Greek letters α and β (and γ, δ, and others) are often used to distinguish two different kinds of subunits within a multisubunit protein, regardless of what kinds of secondary structure may predominate in the subunits. Because hemoglobin is four times as large as myoglobin, much more time and effort were required to solve its three-dimensional structure by x-ray analysis, finally achieved by Max Perutz, John Kendrew, and their colleagues in 1959. The subunits of hemoglobin are arranged in symmetric pairs (Fig. 4-22), each pair having one α subunit and one β subunit. Hemoglobin can therefore be described either as a tetramer or as a dimer of αβ protomers. The role these distinct subunits play in hemoglobin function is discussed extensively in Chapter 5.
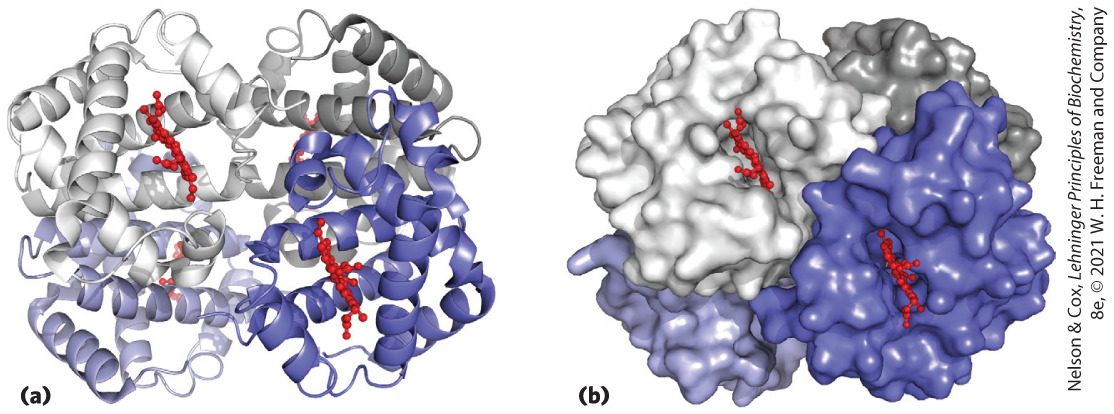
FIGURE 4-22 Quaternary structure of deoxyhemoglobin. X-ray diffraction analysis of deoxyhemoglobin (hemoglobin without oxygen molecules bound to the heme groups) shows how the four polypeptide subunits are packed together. (a) A ribbon representation reveals the secondary structural elements of the structure and the positioning of all the heme prosthetic groups. (b) A surface contour model shows the pockets in which the heme prosthetic groups are bound and helps to visualize subunit packing. The α subunits are shown in shades of gray, the β subunits in shades of blue. Note that the heme groups (red) are relatively far apart. [Data from PDB ID 2HHB, G. Fermi et al., J. Mol. Biol. 175:159, 1984.]

Max Perutz, 1914–2002 (left), and John Kendrew, 1917–1997
SUMMARY 4.3 Protein Tertiary and Quaternary Structures
- Tertiary structure is the complete three-dimensional structure of a polypeptide chain. Many proteins fall into one of four general classes based on tertiary structure: fibrous, globular, membrane, or disordered.
- Insoluble fibrous proteins, such as those that make up keratin, collagen, and silk, have simple repeating elements of secondary structure. In some fibrous proteins, the individual polypeptide chains interact to form complex quaternary structures like coiled coils for strength and flexibility.
- Globular proteins have more complicated tertiary structures, often containing several types of secondary structure in the same polypeptide chain, and fulfill many different functional roles in the cell.
- The first globular protein structure to be determined, by x-ray diffraction methods, was that of the -binding protein myoglobin. The myoglobin structure revealed for the first time how protein structure and function are connected.
- The complex structures of globular proteins can be analyzed by examination of folding patterns, called motifs or folds. The many thousands of known protein structures are generally assembled from a repertoire of only a few hundred motifs. Domains are regions of a polypeptide chain that can fold stably and independently.
- Some proteins or protein segments are intrinsically disordered, lacking definable three-dimensional structure. These proteins often have distinctive amino acid compositions that allow a more flexible structure, which is critical for their biological function.
- Based on structural similarities, proteins can be organized into families and superfamilies, which are informative about protein function and evolution.
- Quaternary structure results from interactions between the subunits of multisubunit (multimeric) proteins or large supramolecular assemblies. Some multimeric proteins are composed of repeated subunits called protomers.
 Tertiary structure is the complete three-dimensional structure of a polypeptide chain. Many proteins fall into one of four general classes based on tertiary structure: fibrous, globular, membrane, or disordered.
Tertiary structure is the complete three-dimensional structure of a polypeptide chain. Many proteins fall into one of four general classes based on tertiary structure: fibrous, globular, membrane, or disordered.