Chapter Review
KEY TERMS
Terms in bold are defined in the glossary.
- conformation
- native conformation
- hydrophobic effect
- solvation layer
- secondary structure
- α helix
- β conformation
- β sheet
- β turn
- Ramachandran plot
- circular dichroism (CD) spectroscopy
- tertiary structure
- quaternary structure
- fibrous proteins
- globular proteins
- intrinsically disordered proteins
- α-keratin
- collagen
- fibroin
- Protein Data Bank (PDB)
- motif
- fold
- domain
- topology diagram
- protein family
- multimer
- oligomer
- protomer
- proteostasis
- denaturation
- renaturation
- chaperone
- Hsp70
- chaperonin
- protein disulfide isomerase (PDI)
- peptide prolyl cis-trans isomerase (PPI)
- amyloid
- amyloidoses
- prion
- x-ray crystallography
- nuclear magnetic resonance (NMR) spectroscopy
- cryo-electron microscopy (cryo-EM)
Problems
1. Properties of the Peptide Bond In x-ray studies of crystalline peptides, Linus Pauling and Robert Corey found that the bond in the peptide link is intermediate in length (1.32 Å) between a typical single bond (1.49 Å) and a double bond (1.27 Å). They also found that the peptide bond is planar (all four atoms attached to the group are located in the same plane) and that the two α-carbon atoms attached to the are always trans to each other (on opposite sides of the peptide bond).
- What does the length of the bond in the peptide linkage indicate about its strength and its bond order (i.e., whether it is single, double, or triple)?
- What do Pauling and Corey’s observations tell us about the ease of rotation about the peptide bond?
2. Structural and Functional Relationships in Fibrous Proteins William Astbury discovered that the x-ray diffraction pattern of wool shows a repeating structural unit spaced about 5.2 Å along the length of the wool fiber. When he steamed and stretched the wool, the x-ray pattern showed a new repeating structural unit at a spacing of 7.0 Å. Steaming and stretching the wool and then letting it shrink gave an x-ray pattern consistent with the original spacing of about 5.2 Å. Although these observations provided important clues to the molecular structure of wool, Astbury was unable to interpret them at the time.
- Given our current understanding of the structure of wool, interpret Astbury’s observations.
- When wool sweaters or socks are washed in hot water or heated in a dryer, they shrink. Silk, on the other hand, does not shrink under the same conditions. Explain.
3. Rate of Synthesis of Hair α-Keratin Hair grows at a rate of 15 to 20 cm/yr. All this growth is concentrated at the base of the hair fiber, where α-keratin filaments are synthesized inside living epidermal cells and assembled into ropelike structures (see Fig. 4-10). The fundamental structural element of α-keratin is the α helix, which has 3.6 amino acid residues per turn and a rise of 5.4 Å per turn (see Fig. 4-3a). Assuming that the biosynthesis of α-helical keratin chains is the rate-limiting factor in the growth of hair, calculate the rate at which peptide bonds of α-keratin chains must be synthesized (peptide bonds per second) to account for the observed yearly growth of hair.
4. Effect of pH on the Conformation of α-Helical Secondary Structures Specific rotation is a measure of a solution’s capacity to rotate circularly polarized light. The unfolding of the α helix of a polypeptide to a randomly coiled conformation is accompanied by a large decrease in a property called specific rotation. Polyglutamate, a polypeptide made up of only l-Glu residues, is an α helix at pH 3. When researchers raise the pH to 7, there is a large decrease in the specific rotation of the solution. Similarly, polylysine (l-Lys residues) is an α helix at pH 10, but when researchers lower the pH to 7, the specific rotation also decreases, as shown in the graph.
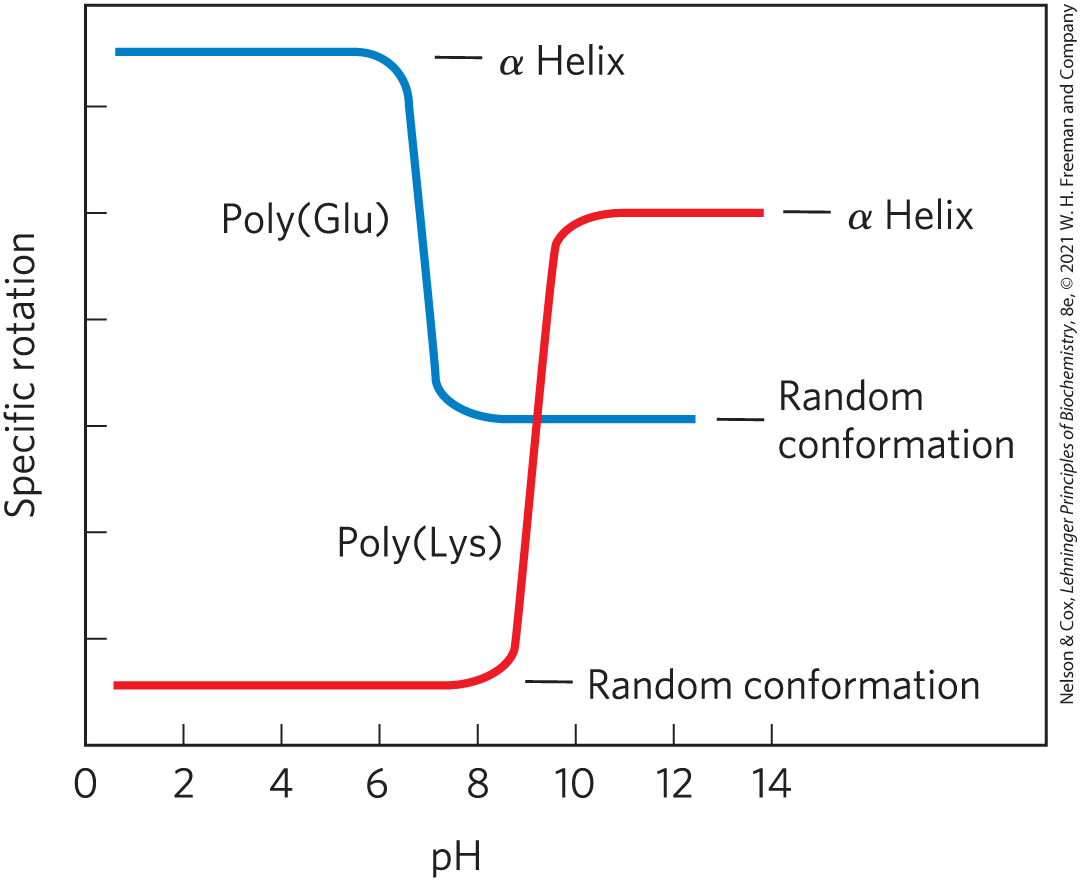
Explain the effect of the pH changes on the conformations of poly(Glu) and poly(Lys). Why does the transition occur over such a narrow range of pH?
5. Disulfide Bonds Determine the Properties of Many Proteins Some natural proteins are rich in disulfide bonds, and their mechanical properties, such as tensile strength, viscosity, and hardness, correlate with the degree of disulfide bonding.
- Glutenin, a wheat protein rich in disulfide bonds, imparts the cohesive and elastic character of dough made from wheat flour. Similarly, the hard, tough nature of tortoise shell results from the extensive disulfide bonding in its α-keratin. What is the molecular basis for the correlation between disulfide-bond content and mechanical properties of the protein?
- Most globular proteins denature and lose their activity when they are briefly heated to . However, the denaturation of globular proteins that contain multiple disulfide bonds often requires longer heat exposure at higher temperatures. One such protein is bovine pancreatic trypsin inhibitor (BPTI), which has 58 amino acid residues in a single peptide chain and contains three disulfide bonds. After a solution of denatured BPTI is cooled, the protein regains its activity. What is the molecular basis for this property of BPTI?
6. Dihedral Angles Consider the series of torsion angles, ϕ and ψ, that might be taken up by the peptide backbone. Which of these closely correspond to ϕ and ψ for an idealized collagen triple helix? Refer to Figure 4-8 as a guide.
7. Amino Acid Sequence and Protein Structure Our growing understanding of how proteins fold allows researchers to make predictions about protein structure based on primary amino acid sequence data. Consider this amino acid sequence.

- Where might bends or β turns occur?
- Where might intrachain disulfide cross-linkages form?
- Suppose that this sequence is part of a larger globular protein. Indicate the probable location (external surface or interior of the protein) of each amino acid residue: Asp, Ile, Thr, Ala, Gln, Lys. Explain your reasoning. (Hint: See the hydropathy index in Table 3-1.)
8. Amino Acid Contributions to Protein Folding Like ribonuclease A, lysozyme from T4 phage is a model enzyme for understanding the energetics and pathways of protein folding. Unlike ribonuclease A, however, T4 lysozyme does not contain any disulfide bonds. A number of studies have quantified the thermodynamic contributions that individual amino acid residues and their interactions make to T4 lysozyme folding.
- An ion pair between an Asp and a His residue in T4 lysozyme contributes 13–21 kJ/mol of favorable folding energy at pH 6.0. However, this ion pair contributes much less to lysozyme folding at either pH 2.0 or pH 10.0. How can you explain this observation?
- Suppose that a Met residue buried in the folded, hydrophobic core of T4 lysozyme is replaced by mutation with a Lys residue. How would the mutation affect a plot of the thermal denaturation of T4 lysozyme at pH 3.0? (See Fig. 4-24a for an example of a thermal denaturation plot.)
- Suppose that the thermal denaturation experiment on the protein with the Met to Lys mutation took place at pH 10.0. Predict whether the mutation would have a greater or a lesser impact on protein stability at pH 10.0 than at pH 3.0. Explain your prediction.
9. Bacteriorhodopsin in Purple Membrane Proteins Under the proper environmental conditions, the salt-loving archaeon Halobacterium halobium synthesizes a membrane protein known as bacteriorhodopsin, which is purple because it contains retinal (see Fig. 10-20). Molecules of this protein aggregate into “purple patches” in the cell membrane. Bacteriorhodopsin acts as a light-activated proton pump that provides energy for cell functions. X-ray analysis of this protein reveals that it consists of seven parallel α-helical segments, each of which traverses the bacterial cell membrane (thickness 45 Å). Calculate the minimum number of amino acid residues necessary for one segment of α helix to traverse the membrane completely. Estimate the fraction of the bacteriorhodopsin protein that is involved in membrane-spanning helices. (Use an average amino acid residue weight of 110.)
10. Conservation of Protein Structure Margaret Oakley Dayhoff originated the idea of protein superfamilies after noticing that proteins with diverse amino acid sequences can have similar tertiary structures. Why can protein structure be more highly conserved than individual amino acid sequences?
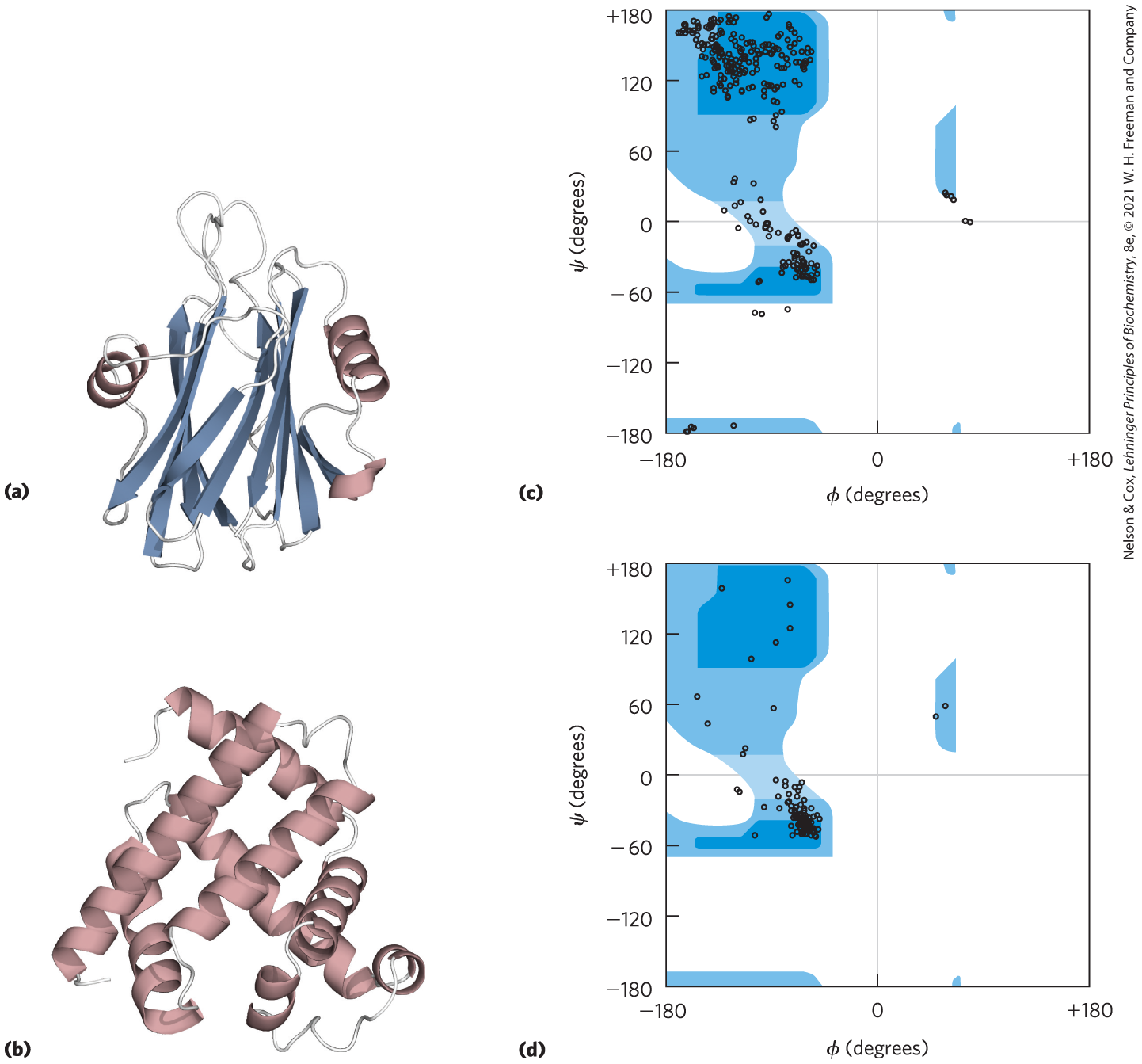
11. Interpreting Ramachandran Plots Examine the two proteins labeled (a) and (b) below. Which of the two Ramachandran plots, labeled (c) and (d) at right, is more likely to be derived from which protein? Why? [Data from (a) PDB ID 1GWY, J. M. Mancheno et al., Structure 11:1319, 2003; (b) PDB ID 1A6M, J. Vojtechovsky et al., Biophys. J. 77:2153, 1999.]
12. Number of Polypeptide Chains in a Multisubunit Protein A researcher treated a sample (660 mg) of an oligomeric protein of with an excess of 1-fluoro-2,4-dinitrobenzene (Sanger’s reagent) under slightly alkaline conditions until the chemical reaction was complete. He then completely hydrolyzed the peptide bonds of the protein by heating it with concentrated HCl. The hydrolysate was found to contain 5.5 mg of the compound shown.

2,4-Dinitrophenyl derivatives of the α-amino groups of other amino acids could not be found.
- Explain how this information can be used to determine the number of polypeptide chains in an oligomeric protein.
- Calculate the number of polypeptide chains in this protein.
- What other analytic technique could you employ to determine whether the polypeptide chains in this protein are similar or different?
13. Amyloid Fibers in Disease Several small aromatic molecules, such as phenol red (used as a nontoxic drug model), have been shown to inhibit the formation of amyloid in laboratory model systems. A goal of the research on these small aromatic compounds is to find a drug that efficiently inhibits the formation of amyloid in the brain in people with incipient Alzheimer disease.
- Suggest why molecules with aromatic substituents would disrupt the formation of amyloid.
- Some researchers suggest that a drug used to treat Alzheimer disease may also be effective in treating type 2 (non-insulin-dependent) diabetes mellitus. Why might a single drug be effective in treating these two different conditions?
14. Protein-Folding Therapies The Food and Drug Administration recently approved the drug lumacaftor for the treatment of cystic fibrosis in patients with the F508ΔCFTR mutation. This mutation is a genetically encoded deletion of amino acid F508 from the protein. About of cystic fibrosis patients have this mutation, and lumacaftor is one of the first drugs that functions as a pharmacological chaperone to correct a defect in the protein-folding process. However, lumacaftor is not always effective in treating patients who have other CFTR mutations that result in misfolding. Why is lumacaftor able to correct the misfolding of some mutant CFTR proteins and not others?
15. Structural Biology Methods Which structural biology method (CD, x-ray crystallography, NMR, or cryo-EM) is best suited to each task?
- Obtaining an ultra-high resolution (<1.5 Å) structure of a drug bound to its protein target
- Obtaining a low-to-medium resolution (5–10 Å) reconstruction of the 11 MDa (11,000,000 Da) bacterial flagellar motor
- Identifying the protonation state and . of a His side chain in an enzyme active site
- Determining whether a protein is intrinsically disordered or contains secondary structure elements
 Amyloid Fibers in Disease Several small aromatic molecules, such as phenol red (used as a nontoxic drug model), have been shown to inhibit the formation of amyloid in laboratory model systems. A goal of the research on these small aromatic compounds is to find a drug that efficiently inhibits the formation of amyloid in the brain in people with incipient Alzheimer disease.
Amyloid Fibers in Disease Several small aromatic molecules, such as phenol red (used as a nontoxic drug model), have been shown to inhibit the formation of amyloid in laboratory model systems. A goal of the research on these small aromatic compounds is to find a drug that efficiently inhibits the formation of amyloid in the brain in people with incipient Alzheimer disease.BIOCHEMISTRY ONLINE
16. Using the PDB The Protein Data Bank (PDB) contains more than 150,000 different three-dimensional biomolecular structures obtained by x-ray crystallography, NMR, and cryo-EM. Each protein structure deposited into the database is given a PDB ID. Several PDB IDs represent proteins whose structures resemble letters from the Roman alphabet. Find each protein structure in the PDB and view the three-dimensional structure using JSmol, PYMOL, or a similar structure viewer.
PDB IDs: 2QYC, 2BNH, 2Q5R, 1XU9, 3H7X, 1OU5, 2WCD
- For each protein, identify its quaternary structure and describe the protomer structure as all α, all β, α/β, or α + β.
- What letter does each protein structure most closely resemble?
- What word(s) can you spell using these protein structures?
17. Protein Modeling Online A group of patients with Crohn disease (an inflammatory bowel disease) underwent biopsies of their intestinal mucosa in an attempt to identify the causative agent. Researchers identified a protein that was present at higher levels in patients with Crohn disease than in patients with an unrelated inflammatory bowel disease or in unaffected controls. The protein was isolated, and the following partial amino acid sequence was obtained (reads left to right):
EAELCPDRCI
HSFQNLGIQC
VKKRDLEQAI
SQRIQTNNNP
FQVPIEEQRG
DYDLNAVRLC
FQVTVRDPSG
RPLRLPPVLP
HPIFDNRAPN
TAELKICRVN
RNSGSCLGGD
EIFLLCDKVQ
KEDIEVYFTG
PGWEARGSFS
QADVHRQVAI
VFRTPPYADP
SLQAPVRVSM
QLRRPSDREL
SEPMEFQYLP
DTDDRHRIEE
KRKRTYETFK
SIMKKSPFSG
PTDPRPPPRR
IAVPSRSSAS
VPKPAPQPYP
- You can identify this protein using a protein database such as UniProt (www.uniprot.org). On the home page, click on the link for a “BLAST” search. On the BLAST page, enter about 30 residues from the protein sequence in the appropriate search field and submit it for analysis. What does this analysis tell you about the identity of the protein?
- Try using different portions of the amino acid sequence. Do you always get the same result?
- A variety of websites provide information about the three-dimensional structure of proteins. Find information about the protein’s secondary, tertiary, and quaternary structures using database sites such as the Protein Data Bank (PDB; www.rcsb.org) or Structural Classification of Proteins (SCOP2; http://scop2.mrc-lmb.cam.ac.uk).
- In the course of your online searches, what did you learn about the cellular function of the protein?
DATA ANALYSIS PROBLEM
18. Mirror-Image Proteins As noted in Chapter 3, “The amino acid residues in protein molecules are almost all l stereoisomers.” It is not clear whether this selectivity is necessary for proper protein function or is an accident of evolution. To explore this question, Milton and colleagues (1992) published a study of an enzyme made entirely of d stereoisomers. The enzyme they chose was HIV protease, a proteolytic enzyme made by HIV that converts inactive viral preproteins to their active forms.
Previously, Wlodawer and coworkers (1989) had reported the complete chemical synthesis of HIV protease from l-amino acids (the l-enzyme), using the process shown in Figure 3-30. Normal HIV protease contains two Cys residues, at positions 67 and 95. Because chemical synthesis of proteins containing Cys is technically difficult, Wlodawer and colleagues substituted the synthetic amino acid l-α-amino-n-butyric acid (Aba) for the two Cys residues in the protein. In the authors’ words, this was done to “reduce synthetic difficulties associated with Cys deprotection and ease product handling.”
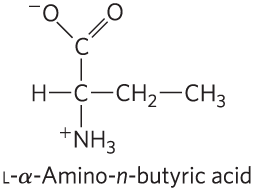
- The structure of Aba is shown below. Why was this a suitable substitution for a Cys residue? Under what circumstances would it not be suitable?
Wlodawer and coworkers denatured the newly synthesized protein by dissolving it in 6 m guanidine HCl and then allowed it to fold slowly by dialyzing away the guanidine against a neutral buffer (10% glycerol, 25mm , pH 7).
- There are many reasons to predict that a protein synthesized, denatured, and folded in this manner would not be active. Give three such reasons.
- Interestingly, the resulting l-protease was active. What does this finding tell you about the role of disulfide bonds in the native HIV protease molecule?
In a more recent study, Milton and coworkers synthesized HIV protease from d-amino acids, using the same protocol as the earlier study (Wlodawer et al.). Formally, there are three possibilities for the folding of the d-protease: it would be (1) the same shape as the l-protease, (2) the mirror image of the l-protease, or (3) something else, possibly inactive.
- For each possibility, decide whether or not it is a likely outcome, and defend your position.
In fact, the d-protease was active: it cleaved a particular synthetic substrate and was inhibited by specific inhibitors. To examine the structure of the d- and l-enzymes, Milton and coworkers tested both forms for activity with d and l forms of a chiral peptide substrate and for inhibition by d and l forms of a chiral peptide-analog inhibitor. Both forms were also tested for inhibition by the achiral inhibitor Evans blue. The findings are given in the table.
Inhibition HIV protease Substrate hydrolysis Peptide inhibitor Evans blue (achiral) d-substrate l-substrate d-inhibitor l-inhibitor l-protease
−
+
−
+
+
d-protease
+
−
+
−
+
- Which of the three models proposed is supported by these data? Explain your reasoning.
- Why does Evans blue inhibit both forms of the protease?
- Would you expect chymotrypsin to digest the d-protease? Explain your reasoning.
- Would you expect total synthesis from d-amino acids followed by renaturation to yield active enzyme for any enzyme? Explain your reasoning.
- The structure of Aba is shown below. Why was this a suitable substitution for a Cys residue? Under what circumstances would it not be suitable?
References
- Milton, R.C., S.C. Milton, and S.B. Kent. 1992. Total chemical synthesis of a d-enzyme: the enantiomers of HIV-1 protease show demonstration of reciprocal chiral substrate specificity. Science 256:1445–1448.
- Wlodawer, A., M. Miller, M. Jaskólski, B.K. Sathyanarayana, E. Baldwin, I.T. Weber, L.M. Selk, L. Clawson, J. Schneider, and S.B. Kent. 1989. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 245:616–621.