Chapter Review
KEY TERMS
Terms in bold are defined in the glossary.
- ammonotelic
- ureotelic
- uricotelic
- aminotransferases
- transaminases
- transamination
- pyridoxal phosphate (PLP)
- oxidative deamination
- l-glutamate dehydrogenase
- glutamine synthetase
- glutaminase
- glucose-alanine cycle
- urea cycle
- urea
- creatine kinase
- essential amino acids
- ketogenic
- glucogenic
- tetrahydrofolate
- S-adenosylmethionine (adoMet)
- tetrahydrobiopterin
- phenylketonuria (PKU)
- mixed-function oxygenases
- alkaptonuria
- maple syrup urine disease
Problems
1. Products of Amino Acid Transamination Name and draw the structure of the α-keto acid resulting when each of the four amino acids listed undergoes transamination with α-ketoglutarate: (a) aspartate, (b) glutamate, (c) alanine, (d) phenylalanine.
2. Measurement of Alanine Aminotransferase Activity The measurement of alanine aminotransferase activity (reaction rate) usually includes an excess of pure lactate dehydrogenase and NADH in the reaction system. The rate of alanine disappearance is equal to the rate of NADH disappearance measured spectrophotometrically. Explain how this assay works.
3. Alanine and Glutamine in the Blood Normal human blood plasma contains all the amino acids required for the synthesis of body proteins, but not in equal concentrations. Alanine and glutamine are present in much higher concentrations than any other amino acids. Suggest why.
4. Glutamate Dehydrogenase Function Increases in ATP levels in blood trigger insulin release by the pancreas, which in turn stimulates the uptake of blood glucose (see Chapter 23, p. 860). Knowing this, suggest why a mutation that prevents inhibition of glutamate dehydrogenase by GTP results in insulin release and hypoglycemia.
5. Distribution of Amino Nitrogen If your diet is rich in alanine but deficient in aspartate, will you show signs of aspartate deficiency? Explain.
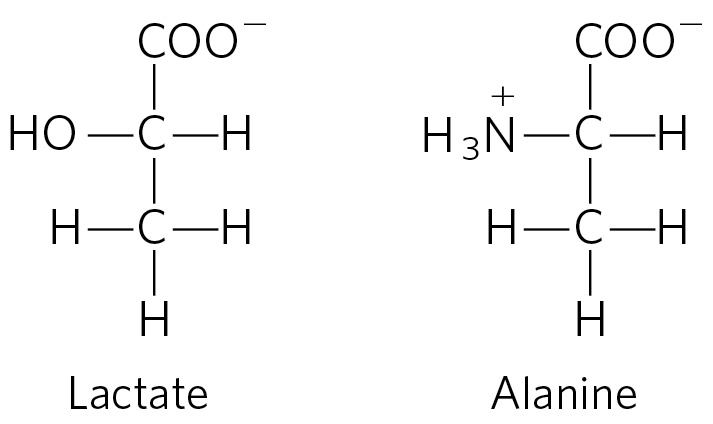
6. Lactate versus Alanine as Metabolic Fuel: The Cost of Nitrogen Removal The three carbons in lactate and alanine have identical oxidation states, and animals can use either carbon source as a metabolic fuel. Compare the net ATP yield (moles of ATP per mole of substrate) for the complete oxidation (to and ) of lactate versus alanine when the cost of nitrogen excretion as urea is included.
7. Ammonia Toxicity Resulting from an Arginine-Deficient Diet In a study, cats were fasted overnight then given a single meal complete in all amino acids except arginine. Within 2 hours, blood ammonia levels increased from a normal level of to , and the cats showed the clinical symptoms of ammonia toxicity. A control group fed a complete amino acid diet or an amino acid diet in which arginine was replaced by ornithine showed no unusual clinical symptoms.
- What was the role of fasting in the experiment?
- What caused the ammonia levels to rise in the experimental group? Why did the absence of arginine lead to ammonia toxicity? Is arginine an essential amino acid in cats? Why or why not?
- Why can ornithine be substituted for arginine?
8. Oxidation of Glutamate Write a series of balanced equations and an overall equation for the net reaction describing the oxidation of 2 mol of glutamate to 2 mol of α-ketoglutarate and 1 mol of urea.
9. Transamination and the Urea Cycle Aspartate aminotransferase has the highest activity of all the mammalian liver aminotransferases. Why?
10. The Case against the Liquid Protein Diet A weight-reducing diet heavily promoted some years ago required the daily intake of a “liquid protein” soup made of hydrolyzed gelatin (derived from collagen), water, and an assortment of vitamins. All other food and drink were to be avoided. People on this diet typically lost 10 to 14 lb in the first week.
- Opponents argued that the weight loss was almost entirely due to water loss and would be regained very soon after a normal diet was resumed. What is the biochemical basis for this argument?
- A few people on this diet died. What are some of the dangers inherent in the diet, and how can they lead to death?
11. Ketogenic Amino Acids Which amino acids are exclusively ketogenic?
12. A Genetic Defect in Amino Acid Metabolism: A Case History A two-year-old child was taken to the hospital. His mother said that he vomited frequently, especially after feedings. The child’s weight and physical development were below normal. His hair, although dark, contained patches of white. A urine sample treated with ferric chloride gave a green color characteristic of the presence of phenylpyruvate. Quantitative analysis of urine samples gave the results shown in the table.
Concentration (mM) Substance Patient’s urine Normal urine Phenylalanine
7.0
0.01
Phenylpyruvate
4.8
0
Phenyllactate
10.3
0
- Suggest which enzyme might be deficient in this child. Propose a treatment.
- Why does phenylalanine appear in the urine in large amounts?
- What is the source of phenylpyruvate and phenyllactate? Why does this pathway (normally not functional) come into play when the concentration of phenylalanine rises?
- Why does the boy’s hair contain patches of white?
13. Role of Cobalamin in Amino Acid Catabolism Pernicious anemia is caused by impaired absorption of vitamin . What is the effect of this impairment on the catabolism of amino acids? Are all amino acids equally affected? (Hint: See Box 17-2.)
14. Vegetarian Diets Vegetarian diets can provide high levels of antioxidants and a lipid profile that can help prevent coronary disease. However, there can be some associated problems. Blood samples were taken from a large group of volunteer subjects who were vegans (strict vegetarians: no animal products), lactovegetarians (vegetarians who eat dairy products), or omnivores (individuals with a varied diet, including meat). In each case, the volunteers had followed the diet for several years. The blood levels of both homocysteine and methylmalonate were elevated in the vegan group, somewhat lower in the lactovegetarian group, and much lower in the omnivore group. Explain.
15. Pernicious Anemia Vitamin deficiency can arise from a few rare genetic diseases that lead to low levels despite a normal diet that includes -rich meat and dairy sources. These conditions cannot be treated with dietary supplements. Explain.
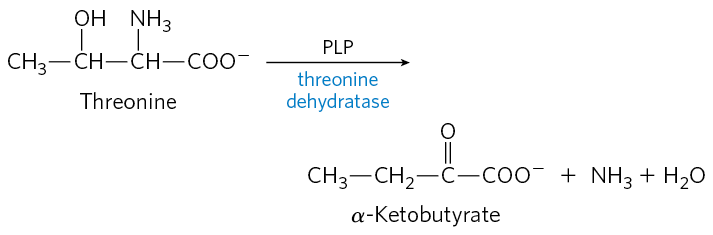
16. Pyridoxal Phosphate Reaction Mechanisms Threonine can be broken down by the enzyme threonine dehydratase, which catalyzes the conversion of threonine to α-ketobutyrate and ammonia. The enzyme uses PLP as a cofactor. Suggest a mechanism for this reaction, based on the mechanisms in Figure 18-6. Note that this reaction includes an elimination at the β carbon of threonine.
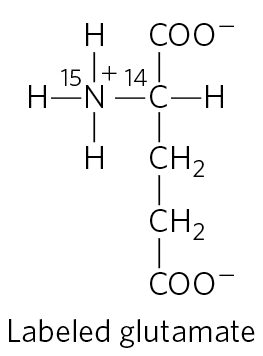
17. Pathway of Carbon and Nitrogen in Glutamate Metabolism When glutamate undergoes oxidative degradation in the liver of a rat, in which atoms of the following metabolites will each isotope be found?
- urea
- succinate
- arginine
- citrulline
- ornithine
- aspartate
18. Chemical Strategy of Isoleucine Catabolism Isoleucine is degraded in six steps to propionyl-CoA and acetyl-CoA.
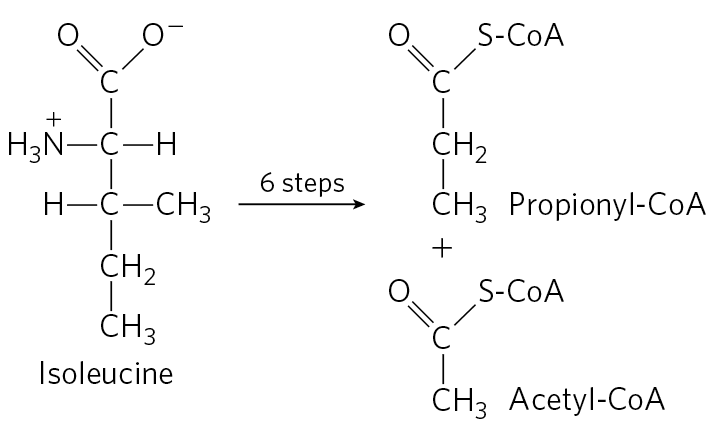
- The chemical process of isoleucine degradation includes strategies analogous to those used in the citric acid cycle and the β oxidation of fatty acids. The intermediates of isoleucine degradation (I to V) shown below are not in the proper order. Use your knowledge and understanding of the citric acid cycle and β-oxidation pathway to arrange the intermediates in the proper metabolic sequence for isoleucine degradation.
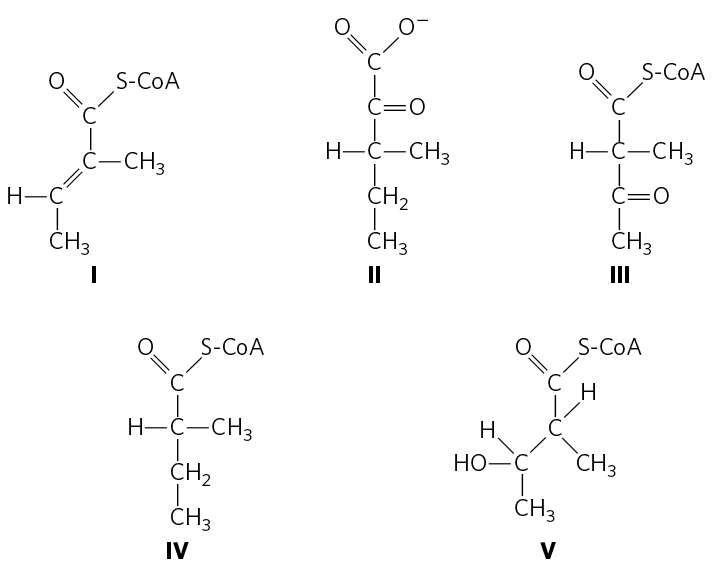
- For each step you propose, describe the chemical process, provide an analogous example from the citric acid cycle or β-oxidation pathway (where possible), and indicate any necessary cofactors.
- The chemical process of isoleucine degradation includes strategies analogous to those used in the citric acid cycle and the β oxidation of fatty acids. The intermediates of isoleucine degradation (I to V) shown below are not in the proper order. Use your knowledge and understanding of the citric acid cycle and β-oxidation pathway to arrange the intermediates in the proper metabolic sequence for isoleucine degradation.
19. Role of Pyridoxal Phosphate in Glycine Metabolism The enzyme serine hydroxymethyltransferase requires pyridoxal phosphate as a cofactor. Propose a mechanism for the reaction catalyzed by this enzyme, in the direction of serine degradation (glycine production). (Hint: See Figs 18-19 and 18-20b.)
20. Parallel Pathways for Amino Acid and Fatty Acid Degradation The carbon skeleton of leucine is degraded by a series of reactions closely analogous to those of the citric acid cycle and β oxidation. For each reaction, (a) through (f), shown below, indicate its type, provide an analogous example from the citric acid cycle or β-oxidation pathway (where possible), and note any necessary cofactors.
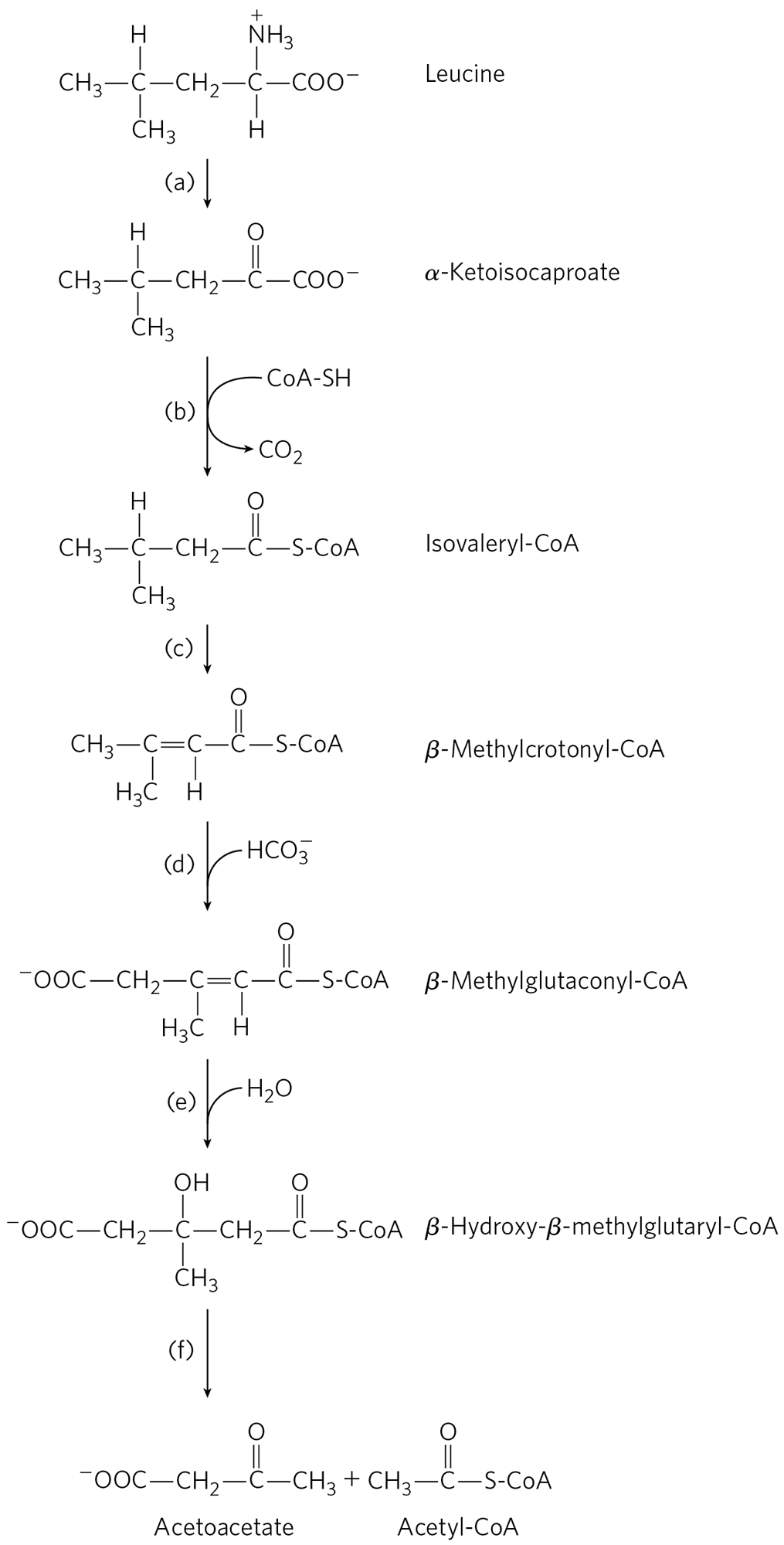
21. Treatments for a Genetic Disease The strict dietary controls required to stem the progress of maple syrup urine disease are difficult to follow for a lifetime, and patients may experience poor metabolic control that leads to neurological symptoms. In these cases, treatment can involve an organ transplant from a suitable donor. Organ transplantation involves considerable risk, but success can greatly alleviate this metabolic disorder and reduce the need for dietary restrictions. Which organ could be transplanted to gain this effect, and why?
 The Case against the Liquid Protein Diet A weight-reducing diet heavily promoted some years ago required the daily intake of a “liquid protein” soup made of hydrolyzed gelatin (derived from collagen), water, and an assortment of vitamins. All other food and drink were to be avoided. People on this diet typically lost 10 to 14 lb in the first week.
The Case against the Liquid Protein Diet A weight-reducing diet heavily promoted some years ago required the daily intake of a “liquid protein” soup made of hydrolyzed gelatin (derived from collagen), water, and an assortment of vitamins. All other food and drink were to be avoided. People on this diet typically lost 10 to 14 lb in the first week.DATA ANALYSIS PROBLEM
22. Maple Syrup Urine Disease Figure 18-28 shows the pathway for the degradation of branched-chain amino acids and the site of the biochemical defect that causes maple syrup urine disease. The initial findings that eventually led to the discovery of the defect in this disease were presented in three papers published in the late 1950s and early 1960s. This problem traces the history of the findings from initial clinical observations to proposal of a biochemical mechanism.
Menkes, Hurst, and Craig (1954) presented the cases of four siblings, all of whom died following a similar course of symptoms. In all four cases, the mother’s pregnancy and the birth had been normal. The first 3 to 5 days of each child’s life were also normal. But soon thereafter each child began having convulsions, and the children died between the ages of 11 days and 3 months. Autopsy showed considerable swelling of the brain in all cases. The children’s urine had a strong, unusual “maple syrup” odor, starting from about the third day of life.
Menkes (1959) reported data collected from six more children. All showed symptoms similar to those described above and died within 15 days to 20 months of birth. In one case, Menkes was able to obtain urine samples during the last months of the infant’s life. When he treated the urine with 2,4-dinitrophenylhydrazone, which forms colored precipitates with keto compounds, he found three α-keto acids in unusually large amounts:
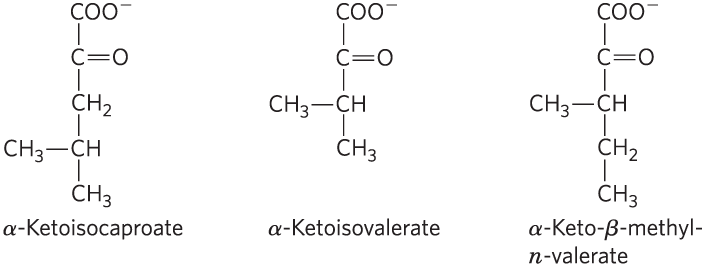
- These α-keto acids are produced by the deamination of amino acids. For each of the α-keto acids above, draw and name the amino acid from which it was derived.
Dancis, Levitz, and Westall (1960) collected further data that led them to propose the biochemical defect shown in Figure 18-28. In one case, they examined a patient whose urine first showed the maple syrup odor when he was 4 months old. At the age of 10 months (March 1956), the child was admitted to the hospital because he had a fever, and he showed grossly delayed motor development. At the age of 20 months (January 1957), he was readmitted and was found to have the degenerative neurological symptoms seen in previous cases of maple syrup urine disease; he died soon after. Results of his blood and urine analyses are shown in the table, along with normal values for each component.
- These α-keto acids are produced by the deamination of amino acids. For each of the α-keto acids above, draw and name the amino acid from which it was derived.
| Urine concentration (mg/24 h) | Plasma concentration (mg/mL) | |||||
|---|---|---|---|---|---|---|
| Normal | Patient | Normal | Patient | |||
| Amino acid(s) | Mar. 1956 | Jan. 1957 | Jan. 1957 | |||
| Alanine | 5–15 | 0.2 | 0.4 | 3.0–4.8 | 0.6 | |
| Asparagine and glutamine | 5–15 | 0.4 | 0 | 3.0–5.0 | 2.0 | |
| Aspartic acid | 1–2 | 0.2 | 1.5 | 0.1–0.2 | 0.04 | |
| Arginine | 1.5–3 | 0.3 | 0.7 | 0.8–1.4 | 0.8 | |
| Cystine | 2–4 | 0.5 | 0.3 | 1.0–1.5 | 0 | |
| Glutamic acid | 1.5–3 | 0.7 | 1.6 | 1.0–1.5 | 0.9 | |
| Glycine | 20–40 | 4.6 | 20.7 | 1.0–2.0 | 1.5 | |
| Histidine | 8–15 | 0.3 | 4.7 | 1.0–1.7 | 0.7 | |
| Isoleucine | 2–5 | 2.0 | 13.5 | 0.8–1.5 | 2.2 | |
| Leucine | 3–8 | 2.7 | 39.4 | 1.7–2.4 | 14.5 | |
| Lysine | 2–12 | 1.6 | 4.3 | 1.5–2.7 | 1.1 | |
| Methionine | 2–5 | 1.4 | 1.4 | 0.3–0.6 | 2.7 | |
| Ornithine | 1–2 | 0 | 1.3 | 0.6–0.8 | 0.5 | |
| Phenylalanine | 2–4 | 0.4 | 2.6 | 1.0–1.7 | 0.8 | |
| Proline | 2–4 | 0.5 | 0.3 | 1.5–3.0 | 0.9 | |
| Serine | 5–15 | 1.2 | 0 | 1.3–2.2 | 0.9 | |
| Taurine | 1–10 | 0.2 | 18.7 | 0.9–1.8 | 0.4 | |
| Threonine | 5–10 | 0.6 | 0 | 1.2–1.6 | 0.3 | |
| Tryptophan | 3–8 | 0.9 | 2.3 | Not measured | 0 | |
| Tyrosine | 4–8 | 0.3 | 3.7 | 1.5–2.3 | 0.7 | |
| Valine | 2–4 | 1.6 | 15.4 | 2.0–3.0 | 13.1 | |
- The table includes taurine, an amino acid not normally found in proteins. Taurine is often produced as a byproduct of cell damage. Its structure is
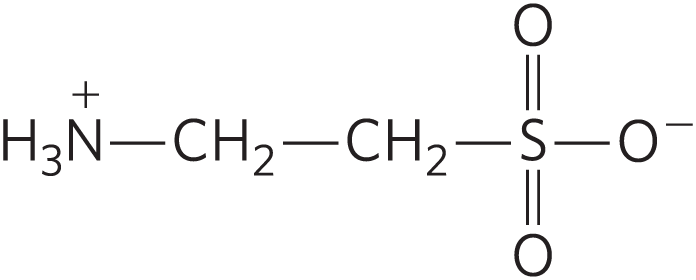
Based on its structure and the information in this chapter, what is the most likely amino acid precursor of taurine? Explain your reasoning.
- Compared with the normal values given in the table, which amino acids showed significantly elevated levels in the patient’s blood in January 1957? Which ones in the patient’s urine?
Based on their results and their knowledge of the pathway shown in Figure 18-28, Dancis and coauthors concluded that “although it appears most likely to the authors that the primary block is in the metabolic degradative pathway of the branched-chain amino acids, this cannot be considered established beyond question.”
- How do the data presented here support this conclusion?
- Which data presented here do not fit this model of maple syrup urine disease? How do you explain these seemingly contradictory data?
- What data would you need to collect to be more secure in your conclusion?
References
- Dancis, J., M. Levitz, and R. Westall. 1960. Maple syrup urine disease: branched-chain ketoaciduria. Pediatrics 25:72–79.
- Menkes, J.H. 1959. Maple syrup disease: isolation and identification of organic acids in the urine. Pediatrics 23:348–353.
- Menkes, J.H., P.L. Hurst, and J.M. Craig. 1954. A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics 14:462–466.