14.2 Feeder Pathways for Glycolysis
Many carbohydrates besides glucose meet their catabolic fate in glycolysis, after being transformed into one of the glycolytic intermediates. The most significant are the storage polysaccharides glycogen and starch, either within cells (endogenous) or obtained in the diet; the disaccharides maltose, lactose, and sucrose; and the monosaccharides fructose, mannose, and galactose (Fig. 14-9).
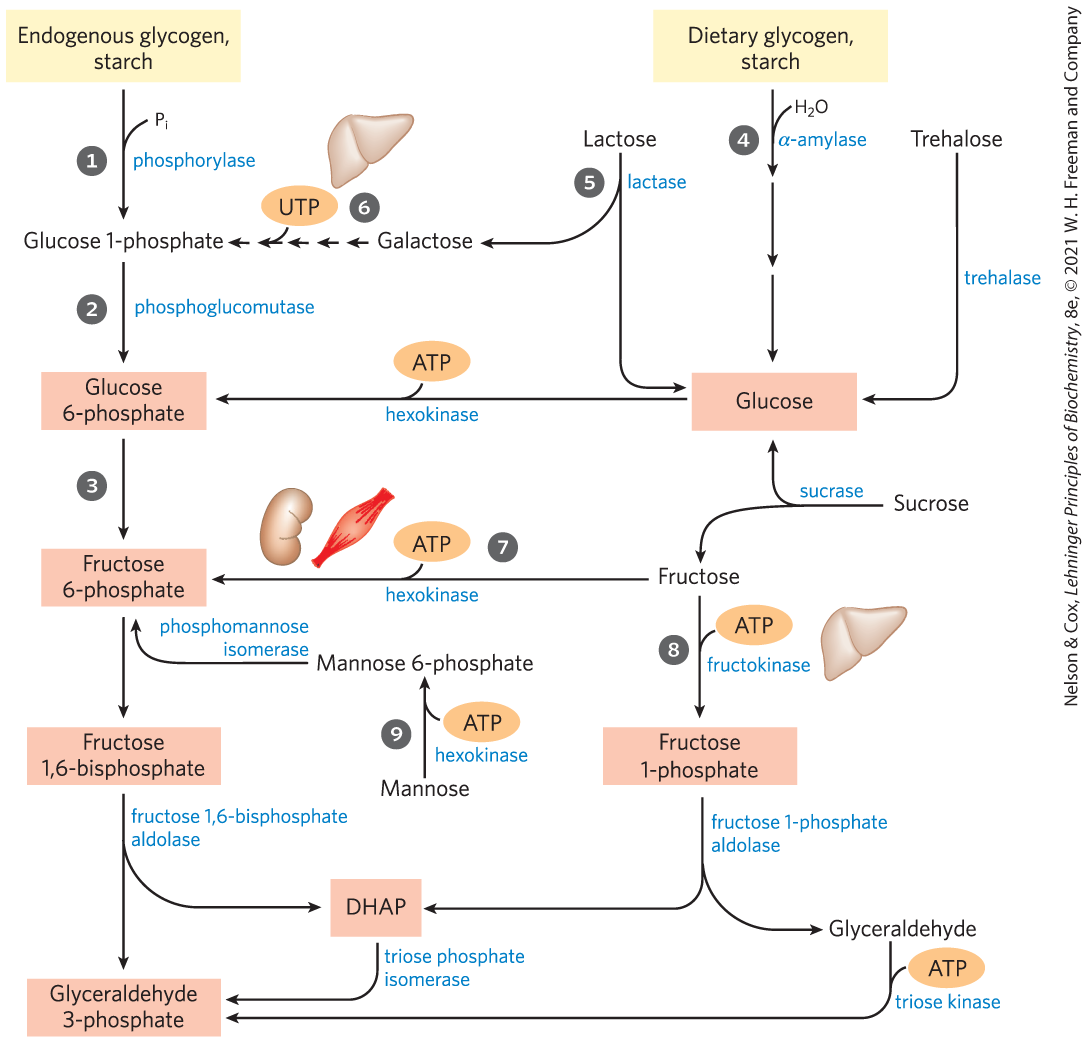
FIGURE 14-9 Entry of dietary glycogen, starch, disaccharides, and hexoses into the preparatory stage of glycolysis. The numbered steps are described in the text.
Endogenous Glycogen and Starch Are Degraded by Phosphorolysis
Glycogen stored in animal tissues (primarily liver and skeletal muscle) and in microorganisms is mobilized for use within the same cell by a phosphorolytic reaction ( in Fig. 14-9) catalyzed by glycogen phosphorylase. The product of this reaction is not free glucose, but glucose 1-phosphate. We discuss glycogen metabolism in more detail in Chapter 15. In plant tissues, starch is mobilized by a similar phosphorolytic reaction catalyzed by starch phosphorylase.
 Glycogen stored in animal tissues (primarily liver and skeletal muscle) and in microorganisms is mobilized for use within the same cell by a phosphorolytic reaction (
Glycogen stored in animal tissues (primarily liver and skeletal muscle) and in microorganisms is mobilized for use within the same cell by a phosphorolytic reaction ( in
in Glucose 1-phosphate produced by glycogen phos-phorylase is converted to glucose 6-phosphate by phosphoglucomutase , which catalyzes the reversible reaction
 , which catalyzes the reversible reaction
, which catalyzes the reversible reactionPhosphoglucomutase employs essentially the same mechanism as phosphoglycerate mutase (Fig. 14-8): both entail a bisphosphate intermediate, and the enzyme is transiently phosphorylated in each catalytic cycle. The general name mutase is given to enzymes that catalyze the transfer of a functional group from one position to another in the same molecule. Mutases are a subclass of isomerases, enzymes that interconvert stereoisomers or structural or positional isomers (see Table 6-3). The glucose 6-phosphate formed in the phosphoglucomutase reaction can continue through glycolysis or enter another pathway such as the pentose phosphate pathway, described in Section 14.6.
 or enter another pathway such as the pentose phosphate pathway, described in
or enter another pathway such as the pentose phosphate pathway, described in WORKED EXAMPLE 14-1 Energy Savings for Glycogen Breakdown by Phosphorolysis
Calculate the energy savings (in ATP molecules per glucose monomer) achieved by breaking down glycogen by phosphorolysis rather than hydrolysis to begin the process of glycolysis.
SOLUTION:
Phosphorolysis produces a phosphorylated glucose (glucose 1-phosphate), which is then converted to glucose 6-phosphate — without expenditure of the cellular energy (1 ATP) needed for formation of glucose 6-phosphate from free glucose. Thus only 1 ATP is consumed per glucose monomer in the preparatory phase, compared with 2 ATP when glycolysis starts with free glucose. The cell therefore gains 3 ATP per glucose monomer (4 ATP produced in the payoff phase minus 1 ATP used in the preparatory phase), rather than 2— a savings of 1 ATP per glucose monomer.
Dietary Polysaccharides and Disaccharides Undergo Hydrolysis to Monosaccharides
For most humans, starch is the major source of carbohydrates in the diet (, Fig. 14-9). Dietary starch has essentially the same structure as glycogen, and its digestion proceeds by the same pathway. Digestion begins in the mouth, where salivary α-amylase hydrolyzes the internal glycosidic linkages of starch and glycogen, producing di- and trisaccharides. These are produced by hydrolysis reactions, in which water, not , is the attacking species. In the stomach, salivary α-amylase is inactivated by the low pH, but a second form of α-amylase, secreted by the pancreas into the small intestine, continues the digestion process.
 ,
, Pancreatic α-amylase release into the small intestine yields mainly maltose and maltotriose (the di- and trisaccharides of glucose) and oligosaccharides called limit dextrins, fragments of amylopectin containing branch points, which are removed by limit dextrinases. Disaccharides are hydrolyzed by a family of membrane-bound hydrolases in the intestinal brush border:

Only monosaccharides are taken up from the intestine. They are actively transported into the intestinal epithelial cells (see Fig. 11-42), then passed into the blood to be carried to various tissues, where they are catabolized via glycolysis.
As we noted in Chapter 7, most animals cannot digest cellulose for lack of the enzyme cellulase, which attacks the glycosidic bonds of cellulose. In ruminant animals, the extended stomach includes a chamber in which symbiotic microorganisms that produce cellulase break down cellulose into glucose molecules. These microorganisms use the resulting glucose in an anaerobic fermentation that produces large quantities of propionate. This propionate, after conversion to succinate (see Fig. 17-12), serves as the starting material for gluconeogenesis, which produces much of the lactose in milk.
Lactose Digestion and Lactose Intolerance
 Lactose Digestion and Lactose Intolerance
Lactose Digestion and Lactose IntoleranceThe defining feature of mammals is, of course, mammary glands, which produce the disaccharide lactose for the nourishment of infants. The enzyme lactase converts lactose to glucose and galactose (, Fig. 14-9), both of which are taken up from the small intestine and metabolized in the tissues by glycolysis. As infants are weaned, their lactase levels diminish, and lactase is absent in most adults — except in certain populations. About one in three adults in northern Europe and in some parts of Africa shows the lactase persistence phenotype. They continue to produce lactase and thus are able to digest milk into adulthood. The other two-thirds experience lactose intolerance due to the disappearance after childhood of most or all of the lactase activity of the intestinal epithelial cells. Without intestinal lactase, lactose cannot be completely digested and absorbed in the small intestine, and it passes into the large intestine, where bacteria convert it to toxic products that cause abdominal cramps and diarrhea. The problem is further complicated because undigested lactose and its metabolites increase the osmolarity of the intestinal contents, favoring retention of water in the intestine, and causing diarrhea. In most parts of the world where lactose intolerance is prevalent, milk is not used as a food by adults, although milk products predigested with lactase are commercially available. In certain human disorders, several or all of the intestinal disaccharidases are missing. In these cases, the digestive disturbances triggered by dietary disaccharides can sometimes be minimized by a controlled diet lacking the undigestible carbohydrates.
 ,
, One way to determine whether lactase is present and active in the intestine is to compare the rise in blood glucose after the ingestion of a quantity of either glucose or lactose. When glucose is ingested, the blood glucose level increases rapidly and transiently. When lactose is ingested, lactase, if present in the intestine, will hydrolyze the lactose into glucose and galactose and the blood glucose level will rise. If lactase is absent or less active, ingesting lactose will lead to little or no transient increase in blood glucose.

Galactose Metabolism and Disease
Galactose (, Fig. 14-9), a product of the hydrolysis of lactose and therefore an important component in the diet of infants, passes in the blood from the intestine to the liver, where it is first phosphorylated at C-1, at the expense of ATP, by the enzyme galactokinase:
 ,
, The galactose 1-phosphate is then transferred to a uridine nucleotide by a transferase. The resulting UDP-galactose is epimerized at C-4, forming UDP-glucose by a set of reactions in which UDP functions as an activator of hexose groups (Fig. 14-10) and a “tag” that these hexoses are in a separate pool from those destined for another process such as glycolysis. The epimerization, catalyzed by UDP-glucose 4-epimerase, involves first the oxidation of the C-4 group to a ketone, then reduction of the ketone to an , with inversion of the configuration at C-4. NAD is the cofactor for both the oxidation and the reduction. The glucose 1-phosphate made this way is converted to glucose 6-phosphate by phosphoglucomutase.
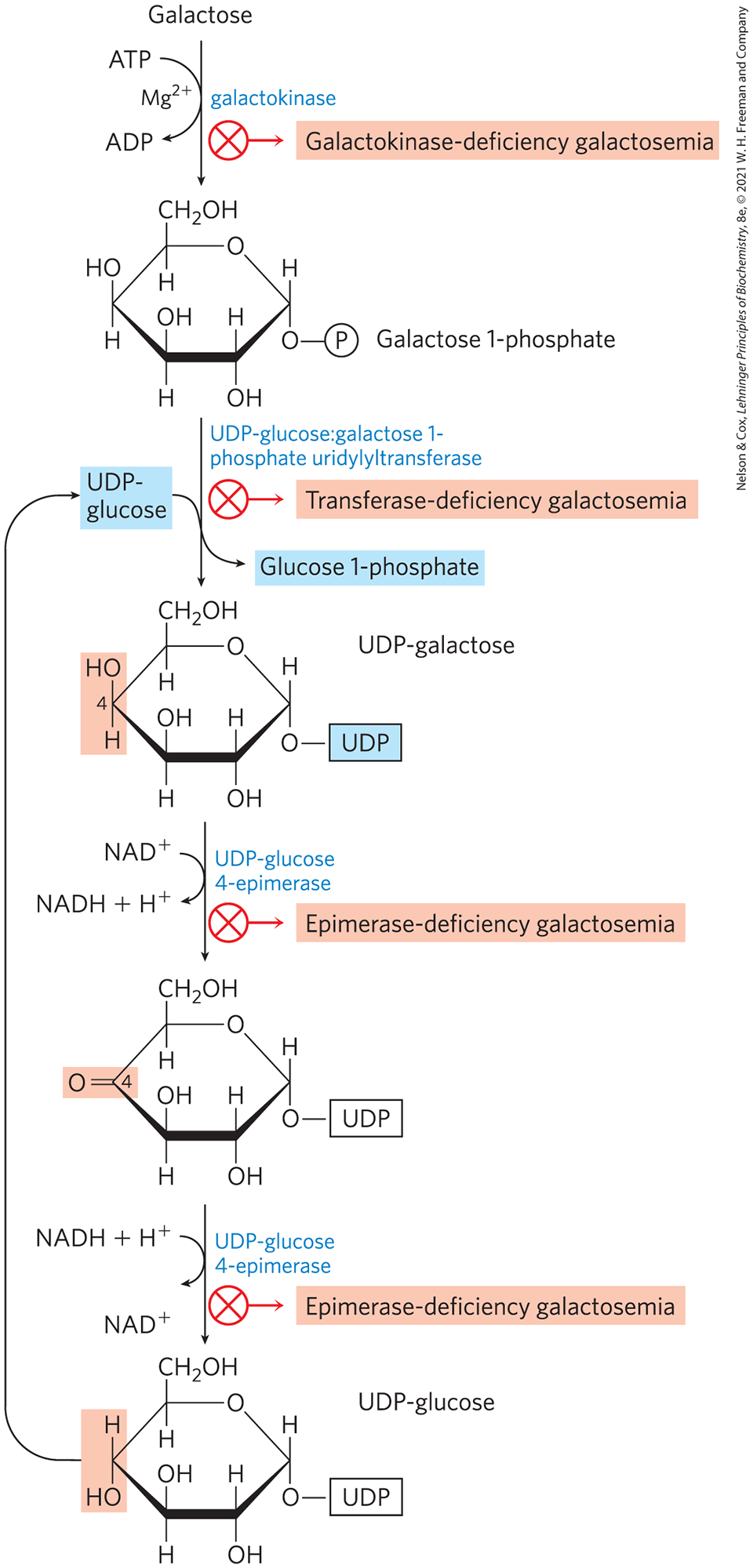
FIGURE 14-10 Conversion of galactose to glucose 1-phosphate. The conversion proceeds through a sugar-nucleotide derivative, UDP-galactose, which is formed when galactose 1-phosphate displaces glucose 1-phosphate from UDP-glucose. UDP-galactose is then converted by UDP-glucose 4-epimerase to UDP-glucose, in a reaction that involves oxidation of C-4 (light red) by , then reduction of C-4 by NADH; the result is inversion of the configuration at C-4. The UDP-glucose is recycled through another round of the same reaction. The net effect of this cycle is the conversion of galactose 1-phosphate to glucose 1-phosphate; there is no net production or consumption of UDP-galactose or UDP-glucose. Defects in the enzymes that catalyze each of these steps result in the various galactosemias shown.
A defect in any of the enzymes in this pathway has serious medical consequences. In galactokinase-deficiency galactosemia, caused by a defect in the GALK gene, high galactose concentrations are found in blood and urine. Affected individuals develop cataracts in infancy, caused by deposition of the galactose metabolite galactitol in the lens.

The other symptoms in this disorder are relatively mild, and strict limitation of galactose in the diet greatly diminishes their severity. Transferase-deficiency galactosemia, caused by a defect in the GALT gene, is more serious; it is characterized by poor growth in childhood, speech abnormality, mental deficiency, and liver damage that may be fatal, even when galactose is withheld from the diet. Epimerase-deficiency galactosemia, caused by a defect in the GALE gene, leads to similar symptoms, but they are less severe when dietary galactose is carefully controlled.
Fructose and Mannose
In most organisms, hexoses other than glucose can undergo glycolysis after conversion to a phosphorylated derivative. Fructose, present in free form in many fruits and formed by hydrolysis of sucrose in the small intestine of vertebrates, is phosphorylated by hexokinase:
This is a major pathway of fructose entry into glycolysis in the muscles and kidney (, Fig. 14-9). In the liver, fructose enters by a different pathway. The liver enzyme fructokinase catalyzes the phosphorylation of fructose at C-1 rather than C-6 (, Fig. 14-9):
 ,
,  ,
, The fructose 1-phosphate is then cleaved to glyceraldehyde and dihydroxyacetone phosphate by fructose 1-phosphate aldolase:
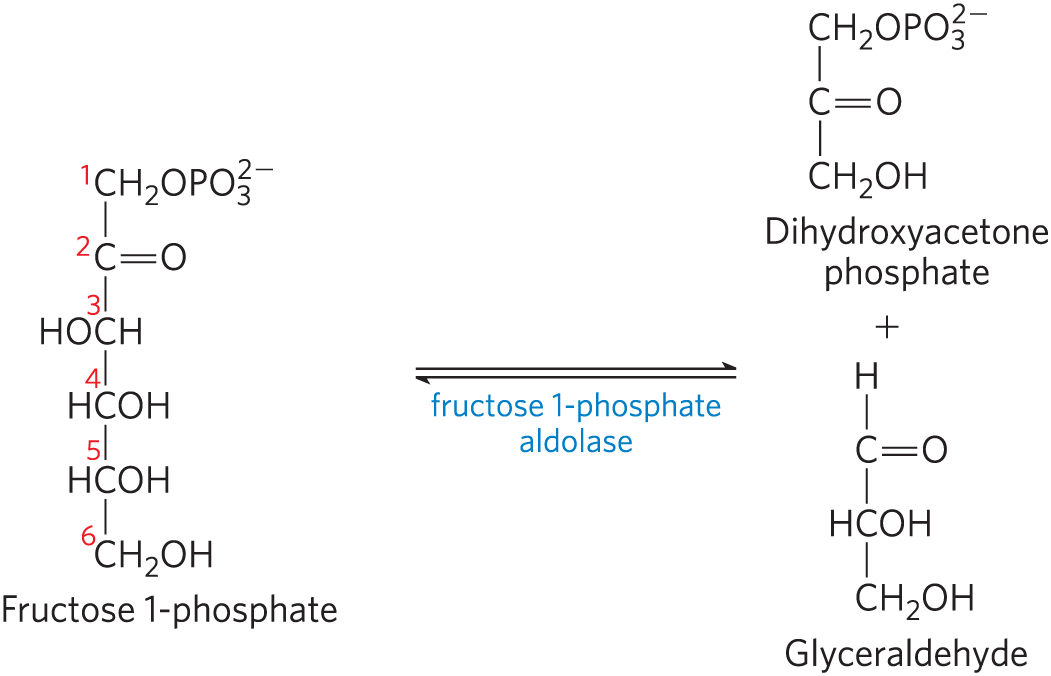
Dihydroxyacetone phosphate is converted to glyceraldehyde 3-phosphate by the glycolytic enzyme triose phosphate isomerase. Glyceraldehyde is phosphorylated by ATP and triose kinase to glyceraldehyde 3-phosphate:
Thus, both products of fructose 1-phosphate hydrolysis enter the glycolytic pathway as glyceraldehyde 3-phosphate.
Mannose, released in the digestion of various polysaccharides and glycoproteins of foods, is phosphorylated at C-6 by hexokinase (, Fig. 14-9):
 ,
,Phosphohexose isomerase converts mannose 6-phosphate to fructose 6-phosphate, which enters glycolysis.
SUMMARY 14.2 Feeder Pathways for Glycolysis
- Endogenous glycogen and starch, polymeric storage forms of glucose, undergo sequential phosphorolysis of glucose residues, forming glucose 1-phosphate. Phosphoglucomutase converts the glucose 1-phosphate to glucose 6-phosphate, which can enter glycolysis at a point in the preparatory phase that requires the investment of only one more ATP.
- Ingested polysaccharides and disaccharides are converted to monosaccharides by hydrolytic enzymes in saliva and in the small intestine. The monosaccharides pass through intestinal cells to the bloodstream, which transports them to the liver or other tissues.
- Lactase is present in infants but often absent in adults, producing lactose intolerance. d-Hexoses, including galactose, fructose, and mannose, can be phosphorylated and funneled into glycolysis. Galactose is converted to glucose 1-phosphate through UDP-galactose and UDP-glucose intermediates. A genetic defect in enzymes of this pathway results in one of several galactosemias of varying severity.
 Endogenous glycogen and starch, polymeric storage forms of glucose, undergo sequential phosphorolysis of glucose residues, forming glucose 1-phosphate. Phosphoglucomutase converts the glucose 1-phosphate to glucose 6-phosphate, which can enter glycolysis at a point in the preparatory phase that requires the investment of only one more ATP.
Endogenous glycogen and starch, polymeric storage forms of glucose, undergo sequential phosphorolysis of glucose residues, forming glucose 1-phosphate. Phosphoglucomutase converts the glucose 1-phosphate to glucose 6-phosphate, which can enter glycolysis at a point in the preparatory phase that requires the investment of only one more ATP.