12.9 Oncogenes, Tumor Suppressor Genes, and Programmed Cell Death
Tumors and cancer are the result of uncontrolled cell division. Normally, cell division is regulated by a family of extracellular growth factors, proteins that cause resting cells to divide and, in some cases, differentiate. The result is a precise balance between the formation of new cells and cell destruction. Regulation of cell division ensures that skin cells are replaced every few weeks and white blood cells are replaced every few days. This is homeostasis at the organismal level. When this balance is disturbed by defects in regulatory proteins, the result is sometimes the formation of a clone of cells that divide repeatedly and without regulation (a tumor) until their presence interferes with the function of normal tissues — cancer. The direct cause is almost always a genetic defect in one or more of the proteins that regulate cell division. In some cases, a defective gene is inherited from one parent; in other cases, the mutation occurs when a toxic compound from the environment (a mutagen or a carcinogen) or high-energy radiation interacts with the DNA of a single cell to damage it and introduce a mutation. In most cases there is both an inherited contribution and an environmental contribution, and in most cases, more than one mutation is required in order to cause completely unregulated division and full-blown cancer.
 When
WhenOncogenes Are Mutant Forms of the Genes for Proteins That Regulate the Cell Cycle
Oncogenes are mutated versions of genes encoding signaling proteins involved in cell cycle regulation. Oncogenes were originally discovered in tumor-causing viruses, then later found to be derived from genes in animal host cells, proto-oncogenes, which encode growth-regulating proteins. During a viral infection, the host DNA sequence of a proto-oncogene is sometimes copied into the viral genome, where it proliferates with the virus. In subsequent viral infection cycles, the proto-oncogenes can become defective by truncation or mutation. Viruses, unlike animal cells, do not have effective mechanisms for correcting mistakes during DNA replication, so they accumulate mutations rapidly. When a virus carrying an oncogene infects a new host cell, the viral DNA (and oncogene) can be incorporated into the host cell’s DNA, where it can now interfere with the regulation of cell division in the host cell. In an alternative, nonviral mechanism, a single cell in a tissue exposed to carcinogens may suffer DNA damage that renders one of its regulatory proteins defective, with the same effect as the viral oncogenic mechanism: failed regulation of cell division.

The mutations that produce oncogenes are genetically dominant; if either of a pair of chromosomes contains a defective gene, that gene product sends the signal “divide,” and a tumor may result. The oncogenic defect can be in any of the proteins involved in communicating the “divide” signal. Oncogenes discovered thus far include those that encode secreted proteins that act as signaling molecules, growth factors, transmembrane proteins (receptors), cytoplasmic proteins (G proteins and protein kinases), and the nuclear transcription factors that control the expression of genes essential for cell division (Jun, Fos).
Some oncogenes encode growth factor receptors with unregulated Tyr kinase activity; they signal continued cell division even when the growth factor is absent, leading to tumor formation. Tumor-producing mutations have been found in many of the signaling protein kinases we have discussed here, all of which use ATP as their substrate for phosphoryl transfer to another element in the signaling cascade. The development of drugs that inhibit the protein kinase activity is an obvious approach to treating cancers that result from unregulated kinase activity. However, most known protein kinase inhibitors act by blocking the binding site for ATP, which is similar in all protein kinases. Inhibitors that are effective against one protein kinase are likely to have intolerable side effects due to their inhibition of other, essential kinases. Nonetheless, the prominent role played by protein kinases in signaling processes related to normal and abnormal cell division has made these enzymes a prime target in the development of drugs for the treatment of cancer (Box 12-4).

Defects in Certain Genes Remove Normal Restraints on Cell Division
Tumor suppressor genes encode proteins that normally restrain cell division. Mutation in one or more of these genes can lead to tumor formation. Unregulated growth due to defective tumor suppressor genes, unlike that due to oncogenes, is genetically recessive; tumors form only if both chromosomes contain a defective gene. This is because the function of these genes is to prevent cell division, and if either copy of the gene is normal, it will produce a normal protein and normal inhibition of division. In a person who inherits one correct copy and one defective copy, every cell begins with one defective copy of the gene. If any one of the individual’s somatic cells undergoes mutation in the one good copy, a tumor may grow from that doubly mutant cell. Mutations in both copies of the genes for pRb, p53, or p21 yield cells in which the normal restraint on cell division is lost and a tumor forms.
Retinoblastoma occurs in children and causes blindness if not surgically treated. The cells of a retinoblastoma have two defective versions of the Rb gene (two defective alleles). Very young children who develop retinoblastoma commonly have multiple tumors in both eyes. These children have inherited one defective copy of the Rb gene, which is present in every cell; each tumor is derived from a single retinal cell that has undergone a mutation in its remaining good copy of the Rb gene. (A fetus with two mutant alleles in every cell is nonviable.) People with retinoblastoma who survive childhood also have a high incidence of cancers of the lung, prostate, and breast later in life.
A far less likely event is that a person born with two good copies of the Rb gene will have independent mutations in both copies in the same cell. Some individuals do develop retinoblastomas later in childhood, usually with only one tumor in one eye. These individuals, presumably, were born with two good copies (alleles) of Rb in every cell, but both Rb alleles in a single retinal cell have undergone mutation, leading to a tumor. After the child reaches about age 3, retinal cells stop dividing, and retinoblastomas at later ages are quite rare.
Stability genes (also called caretaker genes) encode proteins that function in the repair of major genetic defects that result from aberrant DNA replication, ionizing radiation, or environmental carcinogens. Mutations in these genes lead to a high frequency of unrepaired damage (mutations) in other genes, including proto-oncogenes and tumor suppressor genes, and thus to cancer. Among the stability genes are ATM (see Fig. 12-40); the XP gene family, in which mutations lead to xeroderma pigmentosum; and the BRCA1 genes associated with some types of breast cancer (see Box 25-1). Mutations in the gene for p53 also cause tumors; in more than 90% of human cutaneous squamous cell carcinomas (skin cancers) and in about 50% of all other human cancers, p53 is defective. Those very rare individuals who inherit one defective copy of p53 commonly have the Li-Fraumeni cancer syndrome, with multiple cancers (of the breast, brain, bone, blood, lung, and skin) occurring at high frequency and at an early age. The explanation for multiple tumors in this case is the same as that for Rb mutations: an individual born with one defective copy of p53 in every somatic cell is likely to suffer a second p53 mutation in more than one cell during his or her lifetime.
In summary, then, three classes of defects can contribute to the development of cancer: (1) oncogenes, in which the defect is the equivalent of a car’s accelerator pedal being stuck down, with the engine racing; (2) mutated tumor suppressor genes, in which the defect leads to the equivalent of brake failure; and (3) mutated stability genes, with the defect leading to unrepaired damage to the cell’s replication machinery — the equivalent of an unskilled car mechanic.
Mutations in oncogenes and tumor suppressor genes do not have an all-or-none effect. In some cancers, perhaps in all, the progression from a normal cell to a malignant tumor requires an accumulation of mutations (sometimes over several decades), none of which, alone, is responsible for the end effect. For example, the development of colorectal cancer has several recognizable stages, each associated with a mutation (Fig. 12-41). If an epithelial cell in the colon undergoes mutation of both copies of the tumor suppressor gene APC (adenomatous polyposis coli), it begins to divide faster than normal and produces a clone of itself, a benign polyp (early adenoma). For reasons not yet known, the APC mutation results in chromosomal instability, and whole regions of a chromosome are lost or rearranged during cell division. This instability can lead to another mutation, commonly in ras, that converts the clone into an intermediate (precancerous) adenoma.
A third mutation (often in the tumor suppressor gene DCC) leads to a late adenoma. Only when both copies of p53 become defective does this cell mass become a carcinoma — a malignant, life-threatening tumor. The full sequence therefore requires at least seven genetic “hits”: two on each of three tumor suppressor genes (APC, DCCΡ, and p53) and one on the proto-oncogene ras. There are probably several other routes to colorectal cancer as well, but the principle that full malignancy results only from multiple mutations is likely to hold true for all of them. Because mutations accumulate over time, the chances of developing full-blown metastatic cancer rise with age.
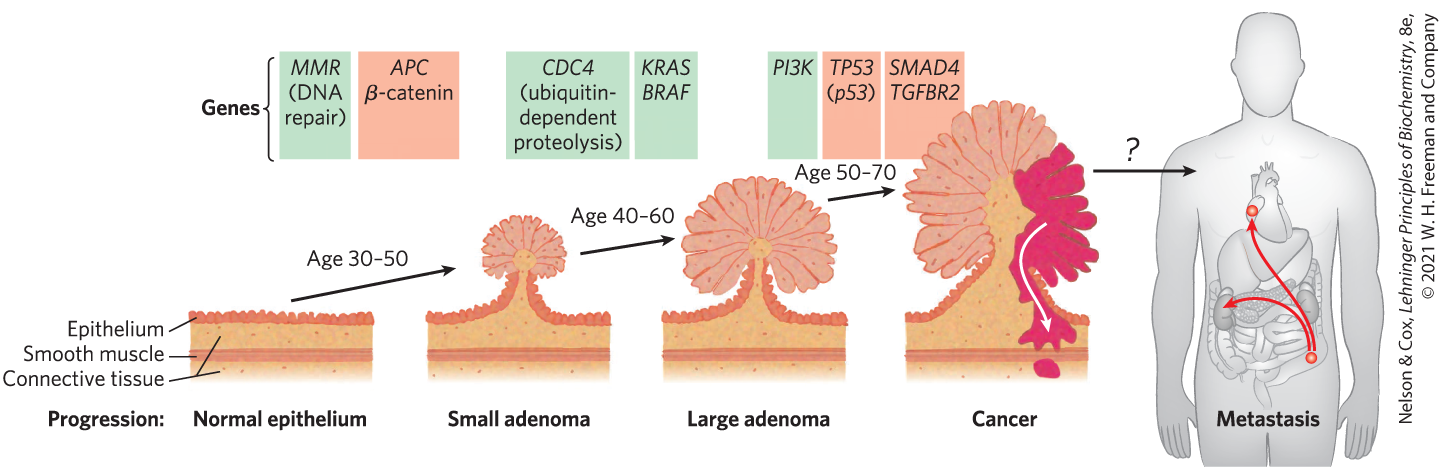
FIGURE 12-41 Multistep transition from normal epithelial cell to colorectal cancer. Serial mutations in oncogenes (green) or tumor suppressor genes (red) lead to progressively less control of cell division, until finally an active tumor forms, which can sometimes metastasize (spread from the initial site to other regions of the body). Mutation of the MMR gene leads to defective DNA repair and consequently to a higher rate of mutation. Mutations in both copies of the tumor suppressor gene APC lead to benign clusters of epithelial cells that multiply too rapidly (early adenoma). The CDC4 oncogene results in defective ubiquitination, which is essential to the regulation of cyclin-dependent kinases (see Fig. 12-38). The oncogenes KRAS and BRAF encode Ras and Raf proteins (see Fig. 12-22), and this further disruption of signaling leads to the formation of a large adenoma, which may be detected by colonoscopy as a benign polyp. Oncogenic mutations in the PI3K gene, which encodes the enzyme phosphoinositide-3 kinase, or in PTEN, which regulates the synthesis of this enzyme, lead to a further strengthening of the signal: divide now. When a cell in one of the polyps undergoes further mutations, such as in the tumor suppressor genes DCC and p53 (see Fig. 12-40), increasingly aggressive tumors form. Finally, mutations in other tumor suppressor genes such as SMAD4 lead to a malignant tumor and sometimes to a metastatic tumor that can spread to other tissues. [Information from S. D. Markowitz and M. M. Bertagnolli, N. Engl. J. Med. 361:2449, 2009, Fig. 2.]
When a polyp is detected in the early adenoma stage and the cells containing the first mutations are removed surgically, late adenomas and carcinomas will not develop; hence the importance of early detection. Cells and organisms, too, have their early detection systems. For example, the ATM and ATR proteins can detect DNA damage too extensive to be repaired effectively. They then trigger, through a pathway that includes p53, the process of apoptosis, in which a cell that has become dangerous to the organism kills itself.
The development of fast and inexpensive sequencing methods has opened a new window on the process by which cancer develops. In a typical study of cancers in humans, the sequences of all 20,000 genes were determined in about 3,300 different tumors, and then compared with the gene sequences in noncancerous tissue from the same patient. Almost 300,000 mutations were detected in all. Only a small fraction of these mutations, the driver mutations, were the cause of unregulated cell division; the vast majority were “passenger mutations,” which occurred randomly and did not confer a selective growth advantage on the tissue in which they occurred. Among the driver mutations were those in about 75 tumor suppressor genes and about 65 oncogenes. These 140 driver mutations fell in three general categories: those that affect cell survival (in genes encoding Ras, PI3K, MAPK, for example), those that affect cells’ ability to maintain an intact genome (ATM, ATR), and those that affect cell fate, causing cells to divide, differentiate, or become quiescent (APC is one example). A relatively small number of mutations were very common in multiple types of cancer, in the genes for Ras, p53, and pRb, for example.
Apoptosis Is Programmed Cell Suicide
Many cells can precisely control the time of their own death by the process of programmed cell death, or apoptosis (pronounced -a--sis; from the Greek for “dropping off,” as in leaves dropping in the fall). One trigger for apoptosis is irreparable damage to DNA. Programmed cell death also occurs during the normal development of an embryo, when some cells must die to give a tissue or an organ its final shape. Carving fingers from stubby limb buds requires the precisely timed death of cells between developing finger bones. During development of the nematode C. elegans from a fertilized egg, exactly 131 cells (of a total of 1,090 somatic cells in the embryo) must undergo programmed death in order to construct the adult body.
Apoptosis also has roles in processes other than development. If a developing antibody-producing cell generates antibodies against a protein or glycoprotein that is normally present in the body, that cell undergoes programmed death in the thymus gland — an essential mechanism for eliminating anti-self antibodies (the cause of many autoimmune diseases). The monthly sloughing of cells of the uterine wall (menstruation) is another case of apoptosis mediating normal cell death. The dropping of leaves in the fall is the result of apoptosis in specific cells of the stem of a plant. Sometimes cell suicide is not programmed but occurs in response to biological circumstances that threaten the rest of the organism. For example, a virus-infected cell that dies before completion of the infection cycle prevents spread of the virus to nearby cells. Severe stresses such as heat, hyperosmolarity, UV light, and gamma irradiation also trigger cell suicide; presumably the organism is better off with any aberrant, potentially mutated cells dead.
The regulatory mechanisms that trigger apoptosis involve some of the same proteins that regulate the cell cycle. The signal for suicide often comes from outside, through a surface receptor. Tumor necrosis factor (TNF), produced by cells of the immune system, interacts with cells through specific TNF receptors. These receptors have TNF-binding sites on the outer face of the plasma membrane and a “death domain” (~80 amino acid residues) that carries the self-destruct signal through the membrane to cytosolic proteins such as TRADD (TNF receptor–associated death domain) (Fig. 12-42).
 The regulatory mechanisms that trigger apoptosis involve some of the same proteins that regulate the cell cycle. The signal for suicide often comes from outside, through a surface receptor. Tumor necrosis factor (TNF), produced by cells of the immune system, interacts with cells through specific TNF receptors. These receptors have TNF-binding sites on the outer face of the plasma membrane and a “death domain” (~80 amino acid residues) that carries the self-destruct signal through the membrane to cytosolic proteins such as TRADD (TNF receptor–associated death domain) (
The regulatory mechanisms that trigger apoptosis involve some of the same proteins that regulate the cell cycle. The signal for suicide often comes from outside, through a surface receptor. Tumor necrosis factor (TNF), produced by cells of the immune system, interacts with cells through specific TNF receptors. These receptors have TNF-binding sites on the outer face of the plasma membrane and a “death domain” (~80 amino acid residues) that carries the self-destruct signal through the membrane to cytosolic proteins such as TRADD (TNF receptor–associated death domain) (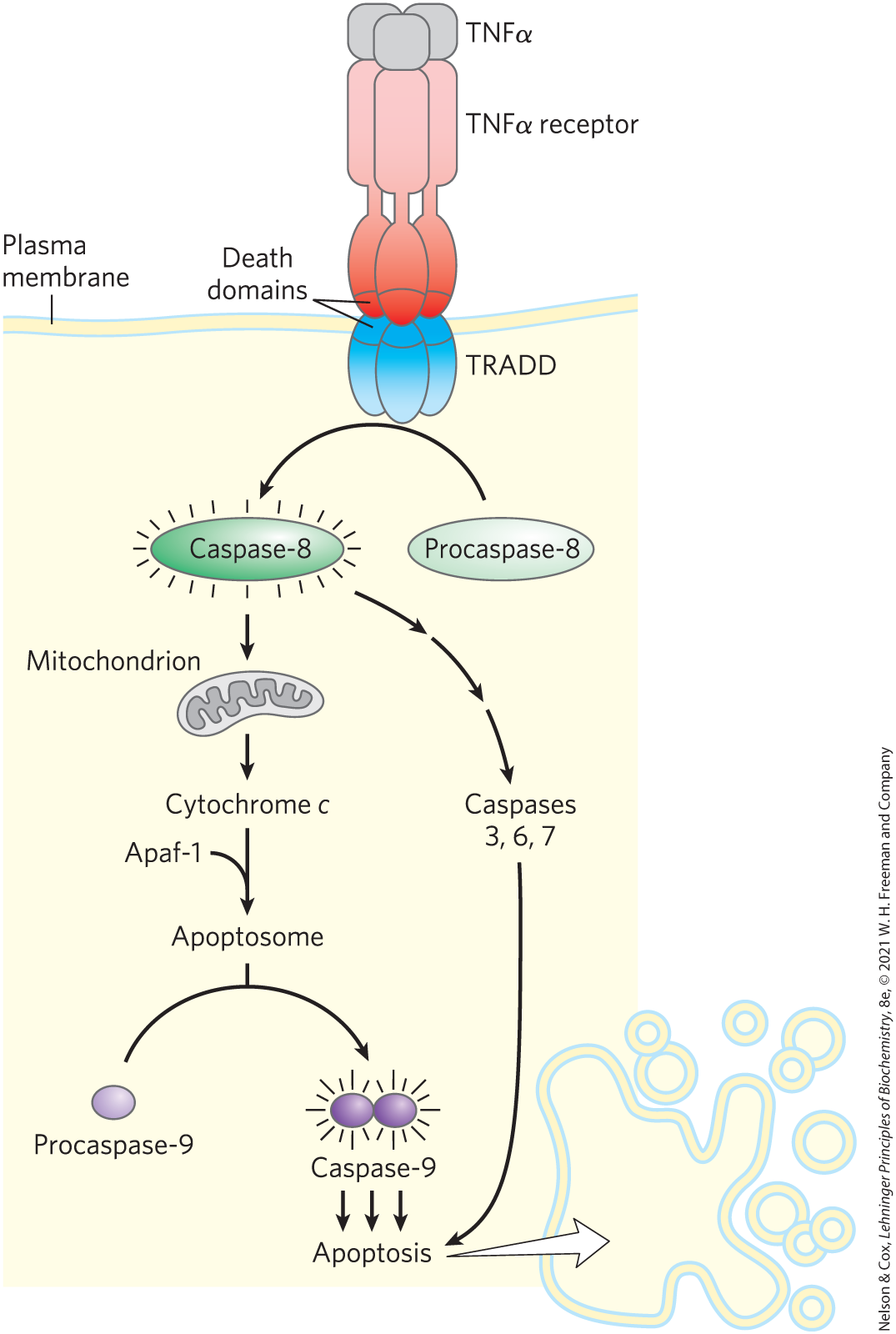
FIGURE 12-42 Initial events of apoptosis. An apoptosis-triggering signal from outside the cell (TNFα) binds to its specific receptor in the plasma membrane. The occupied receptor interacts with the cytosolic protein TRADD through “death domains” (80-residue domains on both TNFα receptor and TRADD), activating TRADD. Activated TRADD initiates a proteolytic cascade that leads to apoptosis: TRADD activates caspase-8, which acts to release cytochrome c from mitochondria, which, in concert with protein Apaf-1, activates caspase-9, triggering apoptosis.
When caspase-8, an “initiator” caspase, is activated by an apoptotic signal carried through TRADD, it further self-activates by cleaving its own proenzyme form. Mitochondria are one target of active caspase-8. The protease causes the release of certain proteins contained between the inner and outer mitochondrial membranes: cytochrome c and several “effector” caspases (see Fig. 19-39). Cytochrome c binds to the proenzyme form of the effector enzyme caspase-9 and stimulates its proteolytic activation. The activated caspase-9, in turn, catalyzes wholesale destruction of cellular proteins — a major cause of apoptotic cell death. One specific target of caspase action is a caspase-activated deoxyribonuclease.
In apoptosis, the monomeric products of protein and DNA degradation (amino acids and nucleotides) are released in a controlled process that allows them to be taken up and reused by neighboring cells. Apoptosis thus allows the organism to eliminate a cell that is unneeded or potentially dangerous without wasting its components.
SUMMARY 12.9 Oncogenes, Tumor Suppressor Genes, and Programmed Cell Death
- Oncogenes encode defective signaling proteins. By continually giving the signal for cell division, they lead to tumor formation. Oncogenes are genetically dominant and may encode defective growth factors, receptors, G proteins, protein kinases, or nuclear regulators of transcription.
- Tumor suppressor genes encode regulatory proteins that normally inhibit cell division; mutations in these genes are genetically recessive but can lead to tumor formation. Cancer is generally the result of an accumulation of mutations in oncogenes and tumor suppressor genes.
- When stability genes, which encode proteins necessary for the repair of genetic damage, are mutated, other mutations go unrepaired, including mutations in proto-oncogenes and tumor suppressor genes that can lead to cancer.
- Apoptosis is programmed and controlled cell death that functions during normal development and adulthood to destroy and recycle unnecessary, damaged, or infected cells. Apoptosis can be triggered by extracellular signals such as TNF, acting through plasma membrane receptors.
 Oncogenes encode defective signaling proteins. By continually giving the signal for cell division, they lead to tumor formation. Oncogenes are genetically dominant and may encode defective growth factors, receptors, G proteins, protein kinases, or nuclear regulators of transcription.
Oncogenes encode defective signaling proteins. By continually giving the signal for cell division, they lead to tumor formation. Oncogenes are genetically dominant and may encode defective growth factors, receptors, G proteins, protein kinases, or nuclear regulators of transcription.