10.4 Working with Lipids
Because lipids are insoluble in water, their extraction and subsequent fractionation require the use of organic solvents and some techniques not commonly used in the purification of water-soluble molecules such as proteins and carbohydrates. In general, complex mixtures of lipids are separated by differences in polarity or solubility in nonpolar solvents. Lipids that contain ester- or amide-linked fatty acids can be hydrolyzed by treatment with acid or alkali or with specific hydrolytic enzymes (phospholipases, glycosidases) to yield their components for analysis. Some methods commonly used in lipid analysis are shown in Figure 10-25 and discussed below.
 In
In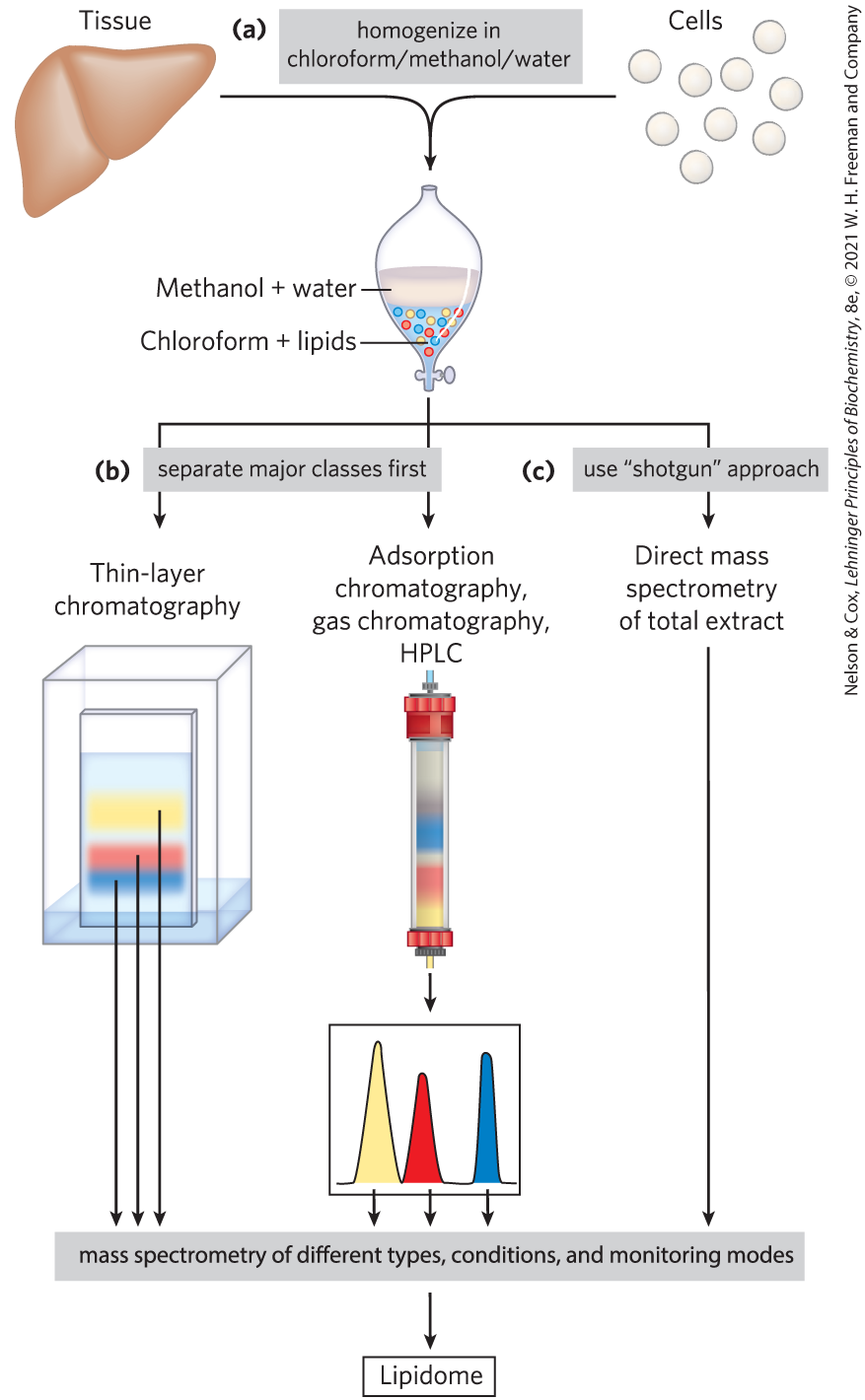
FIGURE 10-25 Common procedures in the extraction, separation, and identification of cellular lipids. (a) Tissue is homogenized in a chloroform/methanol/water mixture, which upon addition of water and removal of unextractable sediment by centrifugation yields two phases. (b) Major classes of extracted lipids in the chloroform phase may first be separated by thin-layer chromatography (TLC), in which lipids are carried up a silica gel–coated plate by a rising solvent front, with less-polar lipids traveling farther than more-polar or charged lipids, or by adsorption chromatography on a column of silica gel, through which solvents of increasing polarity are passed. For example, column chromatography with appropriate solvents can be used to separate closely related lipid species such as phosphatidylserine, phosphatidylglycerol, and phosphatidylinositol. Once separated, each lipid’s complement of fatty acids can be determined by mass spectrometry. (c) Alternatively, in the “shotgun” approach, an unfractionated extract of lipids can be directly subjected to high-resolution mass spectrometry of different types and under different conditions to determine the total composition of all the lipids — that is, the lipidome.
Lipid Extraction Requires Organic Solvents
Neutral lipids (triacylglycerols, waxes, pigments, and so forth) are readily extracted from tissues with ethyl ether, chloroform, or benzene, solvents that do not permit lipid clustering driven by the hydrophobic effect. Membrane lipids are more effectively extracted by more polar organic solvents, such as ethanol or methanol, which reduce the hydrophobic interactions among lipid molecules while also weakening the hydrogen bonds and electrostatic interactions that bind membrane lipids to membrane proteins. A commonly used extractant is a mixture of chloroform, methanol, and water, initially in volume proportions (1:2:0.8) that are miscible, producing a single phase. After tissue is homogenized in this solvent to extract all lipids, more water is added to the resulting extract, and the mixture separates into two phases: methanol/water (top phase) and chloroform (bottom phase). The lipids remain in the chloroform layer, and the more polar molecules such as proteins and sugars partition into the methanol/water layer (Fig. 10-25a).
Adsorption Chromatography Separates Lipids of Different Polarity
Complex mixtures of tissue lipids can be fractionated by chromatographic procedures based on the different polarities of each class of lipid (Fig. 10-25b). In adsorption chromatography, an insoluble, polar material such as silica gel (a form of silicic acid, ) is packed into a glass column, and the lipid mixture (in chloroform solution) is applied to the top of the column. (In high-performance liquid chromatography, or HPLC, the column is of smaller diameter and solvents are forced through the column under high pressure.) The polar lipids bind tightly to the polar silicic acid, but the neutral lipids pass directly through the column and emerge in the first chloroform wash. The polar lipids are then eluted, in order of increasing polarity, by washing the column with solvents of progressively higher polarity. Uncharged but polar lipids (cerebrosides, for example) are eluted with acetone, and very polar or charged lipids (such as glycerophospholipids) are eluted with methanol.
Thin-layer chromatography (TLC) on silicic acid employs the same principle (Fig. 10-25b). A thin layer of silica gel is spread onto a glass plate, to which it adheres. A small sample of lipids dissolved in chloroform is applied near one edge of the plate, which is dipped in a shallow container of an organic solvent or solvent mixture; the entire setup is enclosed in a chamber saturated with the solvent vapor. As the solvent rises on the plate by capillary action, it carries lipids with it. The less polar lipids move farthest, as they have less tendency to bind to the silicic acid. The separated lipids can be detected by spraying the plate with a dye (rhodamine) that fluoresces when associated with lipids, or by exposing the plate to iodine fumes. Iodine reacts reversibly with the double bonds in fatty acids, such that lipids containing unsaturated fatty acids develop a yellow or brown color. Several other spray reagents are also useful in detecting specific lipids. For subsequent analysis, regions containing separated lipids can be scraped from the plate and the lipids recovered by extraction with an organic solvent.
Gas Chromatography Resolves Mixtures of Volatile Lipid Derivatives
Gas chromatography (GC) separates volatile components of a mixture according to their relative tendencies to dissolve in the inert material packed in the chromatography column or to volatilize and move through the column, carried by a current of an inert gas such as helium. Some lipids are naturally volatile, but most must first be derivatized to increase their volatility (that is, lower their boiling point). For an analysis of the fatty acids in a sample of phospholipids, the lipids are first transesterified: heated in a methanol/HCl or methanol/NaOH mixture to convert fatty acids esterified to glycerol into their methyl esters. These fatty acyl methyl esters are then loaded onto the GC column, and the column is heated to volatilize the compounds. Those fatty acyl esters that are most soluble in the column material will partition into (dissolve in) that material; the less-soluble lipids are carried by the stream of inert gas and emerge first from the column. The order of elution depends on the nature of the solid adsorbent in the column and on the boiling point of the components of the lipid mixture. With these techniques, mixtures of fatty acids of various chain lengths and various degrees of unsaturation can be completely resolved.
Specific Hydrolysis Aids in Determination of Lipid Structure
Certain classes of lipids are susceptible to degradation under specific conditions. For example, all ester-linked fatty acids in triacylglycerols, phospholipids, and sterol esters are released by mild acid or alkaline treatment, and somewhat harsher hydrolysis conditions release amide-bound fatty acids from sphingolipids. Enzymes that specifically hydrolyze certain lipids are also useful in the determination of lipid structure. Phospholipases A, C, and D (Fig. 10-14) each split particular bonds in phospholipids and yield products with characteristic solubilities and chromatographic behaviors. Phospholipase C, for example, releases a water-soluble phosphoryl alcohol (such as phosphocholine from phosphatidylcholine) and a chloroform-soluble diacylglycerol, each of which can be characterized separately to determine the structure of the intact phospholipid. The combination of specific hydrolysis with characterization of the products by TLC, GC, or HPLC often allows determination of a lipid structure.
Mass Spectrometry Reveals Complete Lipid Structure
To establish unambiguously the length of a hydrocarbon chain or the position of double bonds, mass spectrometric analysis of lipids or their volatile derivatives is invaluable. The chemical properties of similar lipids (for example, two fatty acids of similar length unsaturated at different positions, or two isoprenoids with different numbers of isoprene units) are very much alike, and their order of elution from the various chromatographic procedures often does not distinguish between them. When the eluate from a chromatography column is sampled by mass spectrometry, however, the components of a lipid mixture can be simultaneously separated and identified by their unique pattern of fragmentation (Fig. 10-26). With the increased resolution of mass spectrometry, it is possible to identify individual lipids in very complex mixtures without first fractionating the lipids in a crude extract. This “shotgun” method (Fig. 10-25c) avoids losses during the preliminary separation of lipid subclasses, and it is faster.
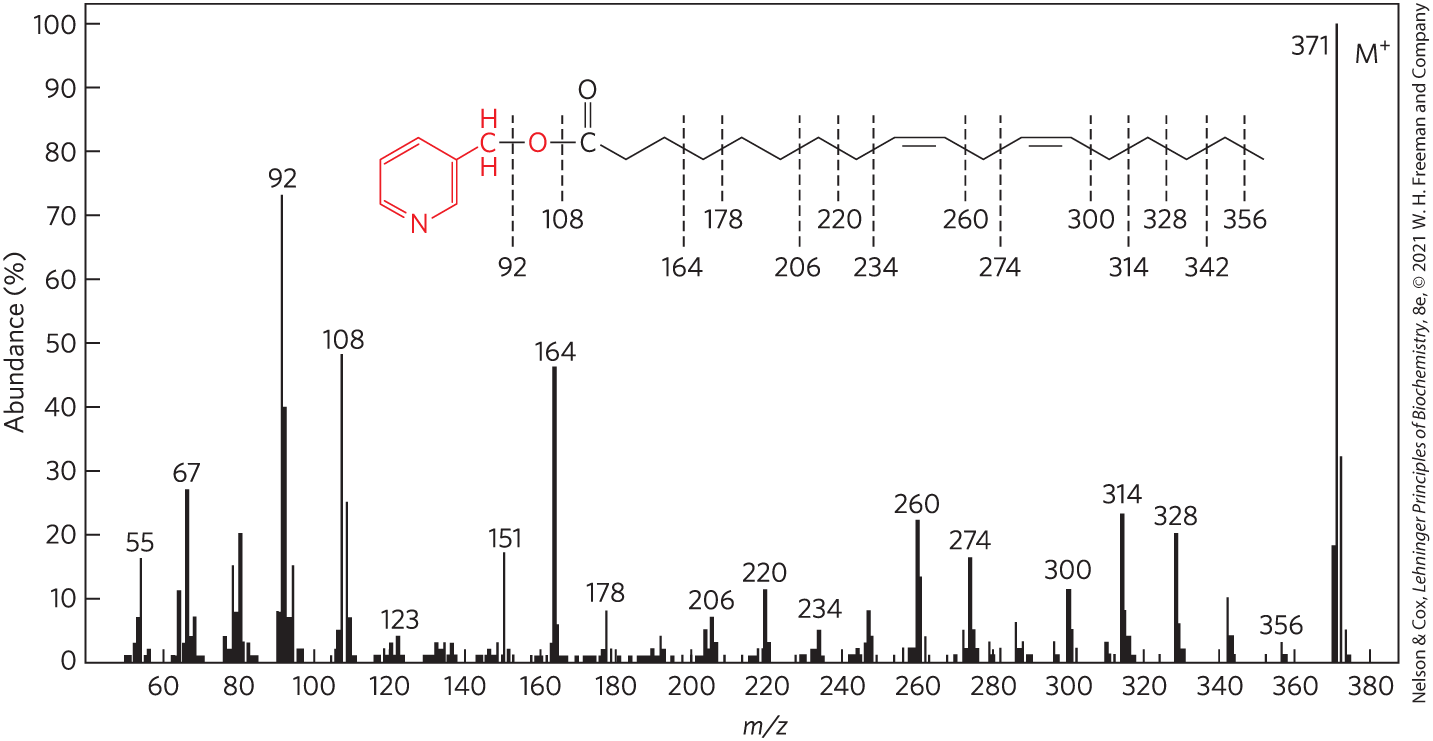
FIGURE 10-26 Determination of fatty acid structure by mass spectrometry. The fatty acid is first converted to a derivative that minimizes migration of the double bonds when the molecule is fragmented by electron bombardment. The derivative shown here is a picolinyl ester of linoleic acid — 18:2() ( 371) — in which the alcohol is picolinol (red). When bombarded with a stream of electrons, this molecule is volatilized and converted to a parent ion (; 371), in which the N atom bears the positive charge, and a series of smaller fragments produced by breakage of bonds in the fatty acid. The mass spectrometer separates these charged fragments according to their mass/charge ratio ().
The prominent ions at and contain the pyridine ring of the picolinol and various fragments of the carboxyl group, showing that the compound is indeed a picolinyl ester. The molecular ion, (), confirms the presence of a fatty acid with two double bonds. The uniform series of ions 14 atomic mass units (u) apart represents loss of each successive methyl and methylene group from the methyl end of the acyl chain (beginning at C-18; the right end of the molecule as shown here), until the ion at is reached. This is followed by a gap of 26 u for the carbons of the terminal double bond, at ; a further gap of 14 u for the C-11 methylene group, at ; and so forth. By this means, the entire structure is determined, although these data alone do not reveal the configuration (cis or trans) of the double bonds. [Information from W. W. Christie, Lipid Technol. 8:64, 1996.]
Lipidomics Seeks to Catalog All Lipids and Their Functions
As lipid biochemists have become aware of the thousands of different naturally occurring lipids, they have created a database analogous to the Protein Data Bank. The LIPID MAPS Lipidomics Gateway (www.lipidmaps.org) has its own classification system that places each lipid species in one of eight chemical categories, each designated by two letters (Table 10-2). Within each category, finer distinctions are indicated by numbered classes and subclasses. For example, all glycerophosphocholines are GP01. The subgroup of glycerophosphocholines with two fatty acids in ester linkage is designated GP0101; the subgroup with one fatty acid ether-linked at position 1 and one ester-linked at position 2 is GP0102. The specific fatty acids are designated by numbers that give every lipid its own unique identifier, so that each individual lipid, including lipid types not yet discovered, can be unambiguously described in terms of a 12-character identifier, the LM_ID. One factor used in this classification system is the nature of the biosynthetic precursor. For example, prenol lipids (such as dolichols and vitamins E and K) are formed from isoprenyl precursors.
| Category | Category code | Examples |
|---|---|---|
Fatty acids |
FA |
Oleate, stearoyl-CoA, palmitoylcarnitine |
Glycerolipids |
GL |
Di- and triacylglycerols |
Glycerophospholipids |
GP |
Phosphatidylcholine, phosphatidylserine, phosphatidyethanoloamine |
Sphingolipids |
SP |
Sphingomyelin, ganglioside GM2 |
Sterol lipids |
ST |
Cholesterol, progesterone, bile acids |
Prenol lipids |
PR |
Farnesol, geraniol, retinol, ubiquinone |
Saccharolipids |
SL |
Lipopolysaccharide |
Polyketides |
PK |
Tetracycline, erythromycin, aflatoxin |
The eight chemical categories in Table 10-2 do not coincide perfectly with the less formal categorization according to biological function that we have used in this chapter. For example, the structural lipids of membranes include both glycerophospholipids and sphingolipids, which are separate categories in Table 10-2. Each method of classification has its advantages.
The application of mass spectrometric techniques with high throughput and high resolution can provide quantitative catalogs of all the lipids present in a specific cell type under particular conditions — the lipidome — and of the ways in which the lipidome changes with differentiation, disease such as cancer, or drug treatment. An animal cell contains more than a thousand different lipid species, each presumably having a specific function. These functions are known for a growing number of lipids, but the still largely unexplored lipidome offers a rich source of new problems for the next generation of biochemists and cell biologists to solve.
SUMMARY 10.4 Working with Lipids
- In the determination of lipid composition, lipids can first be extracted from tissues with organic solvents.
- Lipids in mixtures can be separated on the basis of their polarity and interactions with polar materials such as silica, using adsorption chromatography methods such as HPLC or TLC.
- GC volatilizes lipids so that they can be carried by a stream of inert gas and resolved based on their ability to partition into a soluble column material.
- Phospholipases specific for one of the bonds in a phospholipid can be used to generate simpler compounds for subsequent analysis.
- High-resolution mass spectrometry allows the analysis of crude mixtures of lipids without prefractionation — the “shotgun” approach.
- Lipidomics combines powerful analytical techniques to determine the full complement of lipids in a cell or a tissue (the lipidome) and to assemble annotated databases that allow comparisons between lipids of different cell types and under different conditions.
 In the determination of lipid composition, lipids can first be extracted from tissues with organic solvents.
In the determination of lipid composition, lipids can first be extracted from tissues with organic solvents.