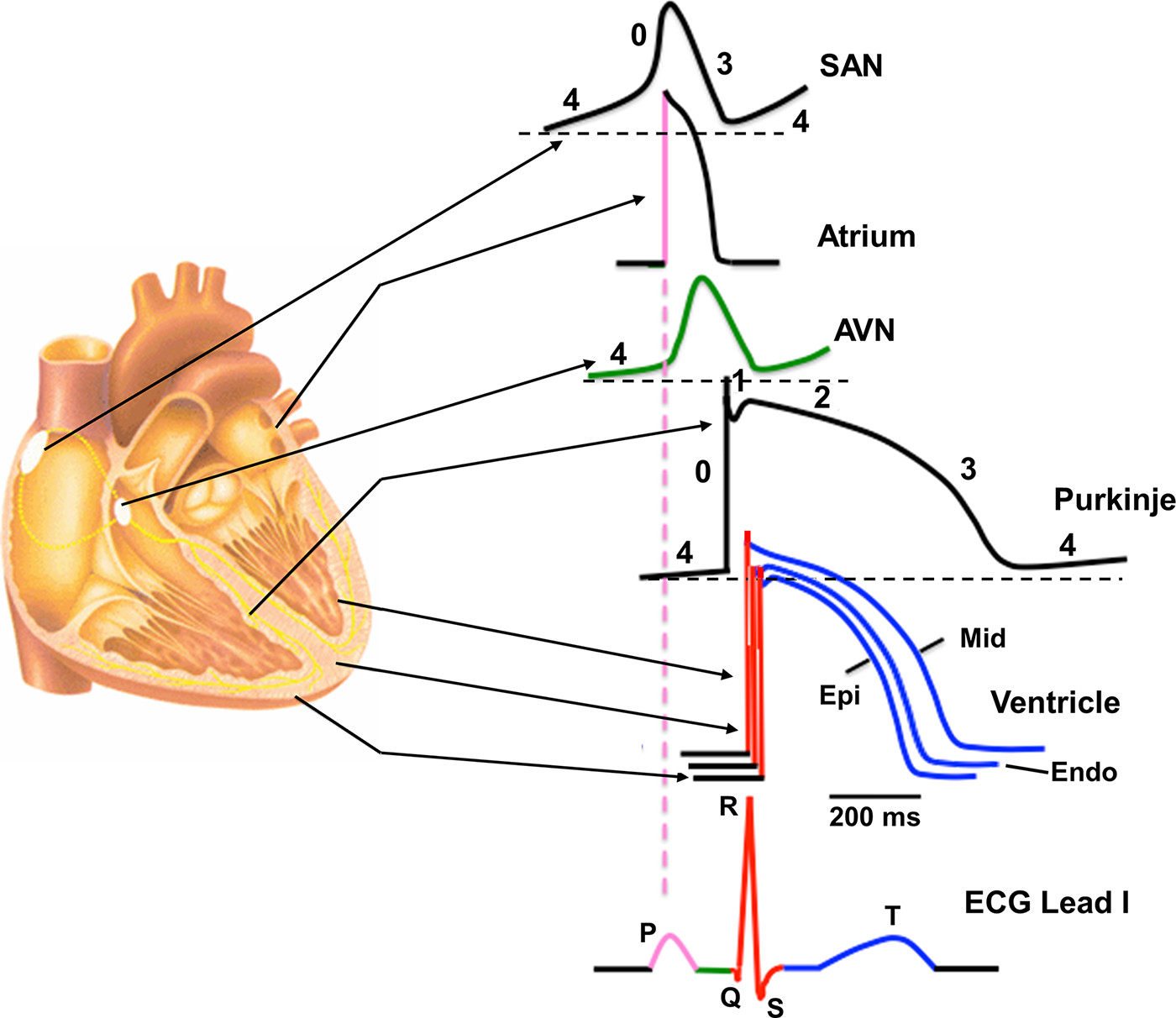
Figure 1 : Regional differences in cardiac action potential configurations. Schematic cross section of the heart depicting the corresponding action potential configuration from different regions of the heart indicated by arrows. Color-coded sections on the action potentials refer to the corresponding sections on the schematic electrocardiogram (ECG). AVN, atrioventricular node; Endo, endocardial; Epi, epicardial; Mid, midmyocardial; SAN, sinoatrial node.
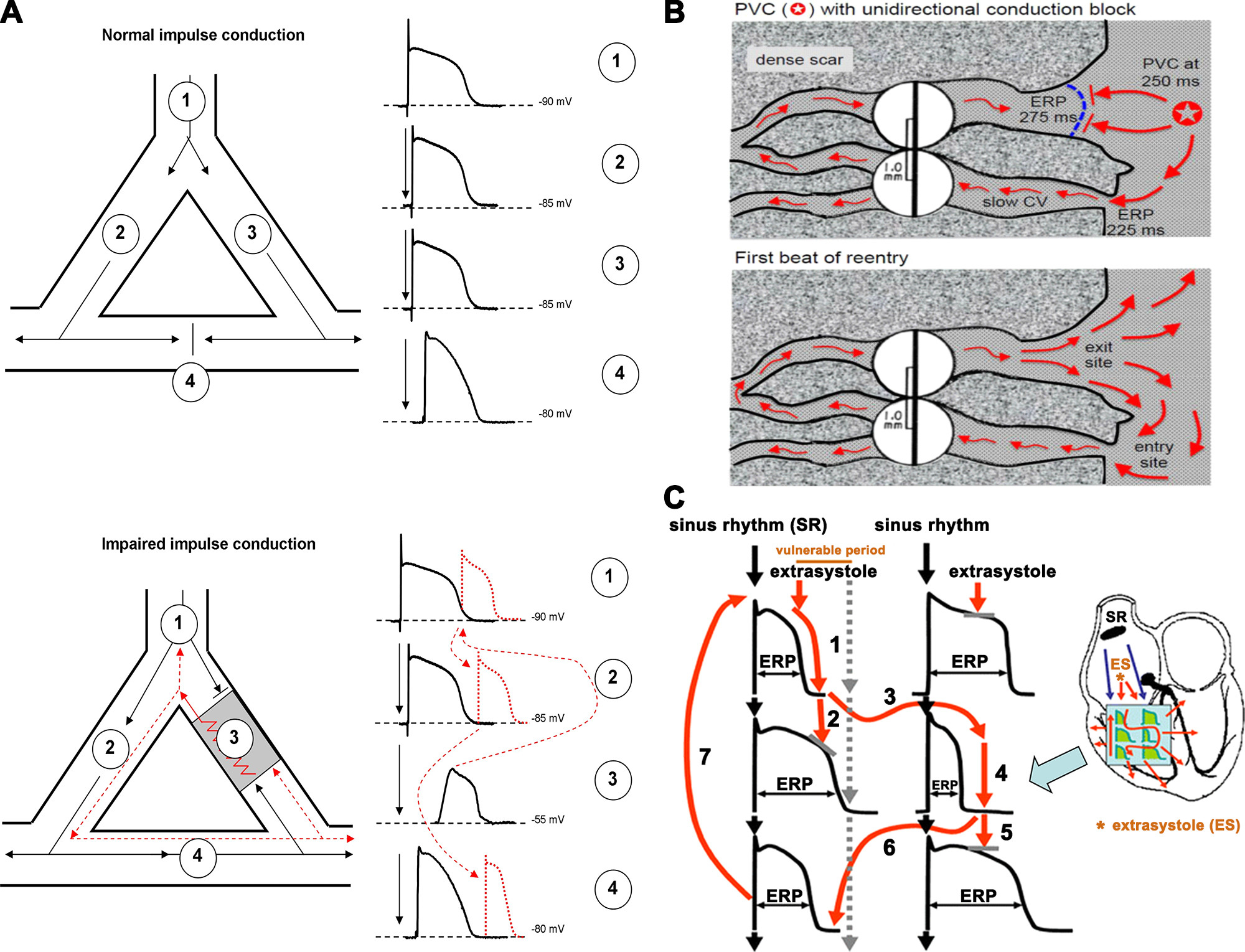
Figure 2 : A, top: an area of branching cardiac tissue, providing separate paths for impulse propagation from proximal (1) to distal (2–4) directions. The separate paths for impulse conduction can be variable, as branching Purkinje fiber-ventricular junctions, ventricular muscle segments with nonconducting fibrosis, scar, or infarcted tissue in the core. If the tissues in these paths are healthy, the impulse conduction is fast and, because of the relatively long effective refractory period (ERP) in cardiac cells, impulses would collide at site 4 and would propagate only into the distal directions. Bottom: an area of depolarized myocardium at site 3, due to asymmetric severe local myocardial damage, imposing conduction block from the anterograde direction. However, in the case that the impulse travels from site 4 in the retrograde direction, it can propagate back very slowly into this damaged area, and if its propagation is slow enough to outlast the ERP in front the impulse can reexcite the proximal tissue. Then, this impulse would propagate toward both sites 1 and 2, establishing a circus movement and reentry arrhythmia. B: example of a classic mechanism by which a premature ventricular complex (PVC) initiates reentry in the fibrotic border zone of an infarct, due to slow conduction and dispersion of refractoriness. Top: a PVC occurring 250 ms after the previous beat arrives too early to propagate through the upper myocyte strand with a long ERP of 275 ms, but it propagates successfully (red arrows) through the lower strand with a shorter ERP of 225 ms (entry site). The impulse propagates slowly (slow CV), eventually reaching the upper strand from the opposite direction. Bottom: if the total conduction time is >275 ms, the interface of the upper strand with normal tissue (exit site) has recovered excitability, and the impulse can then propagate through the region of prior conduction block, thus initiating reentry. Dispersion of refractoriness is caused by electrical remodeling. The slow propagation is due to zig-zag conduction through the myocyte strands as well as gap junction remodeling. (Reproduced from Ref. 17 with permission.) C: schematic illustration of functional reentry mechanism without a well-defined anatomical obstacle. The arrhythmia substrate is represented by artificially enhanced action potential duration differences. In normal circumstances, impulses originating from the sinus node (black arrows) use physiological pathways to propagate through atrial and ventricular tissue and the conduction system. An early ectopic impulse (trigger, red arrow) can only propagate via pathways where the tissue is not depolarized, and consequently its refractoriness is over, whereas the conduction is blocked in directions where the tissue is not fully repolarized and cells are still in the refractory state. Thus the abnormal impulse can travel in a zig-zag direction through reentry paths created by heterogeneous repolarization and conduction. The dispersion of repolarization creates a time window called the vulnerable period, where extra stimuli could elicit the reentry arrhythmia. However, outside this window extra stimuli would only cause a single or multiple relatively harmless extrasystoles. CV, conduction velocity; ES, extrasystole; SR, sinus rhythm. Reproduced from Ref. 18 with permission.
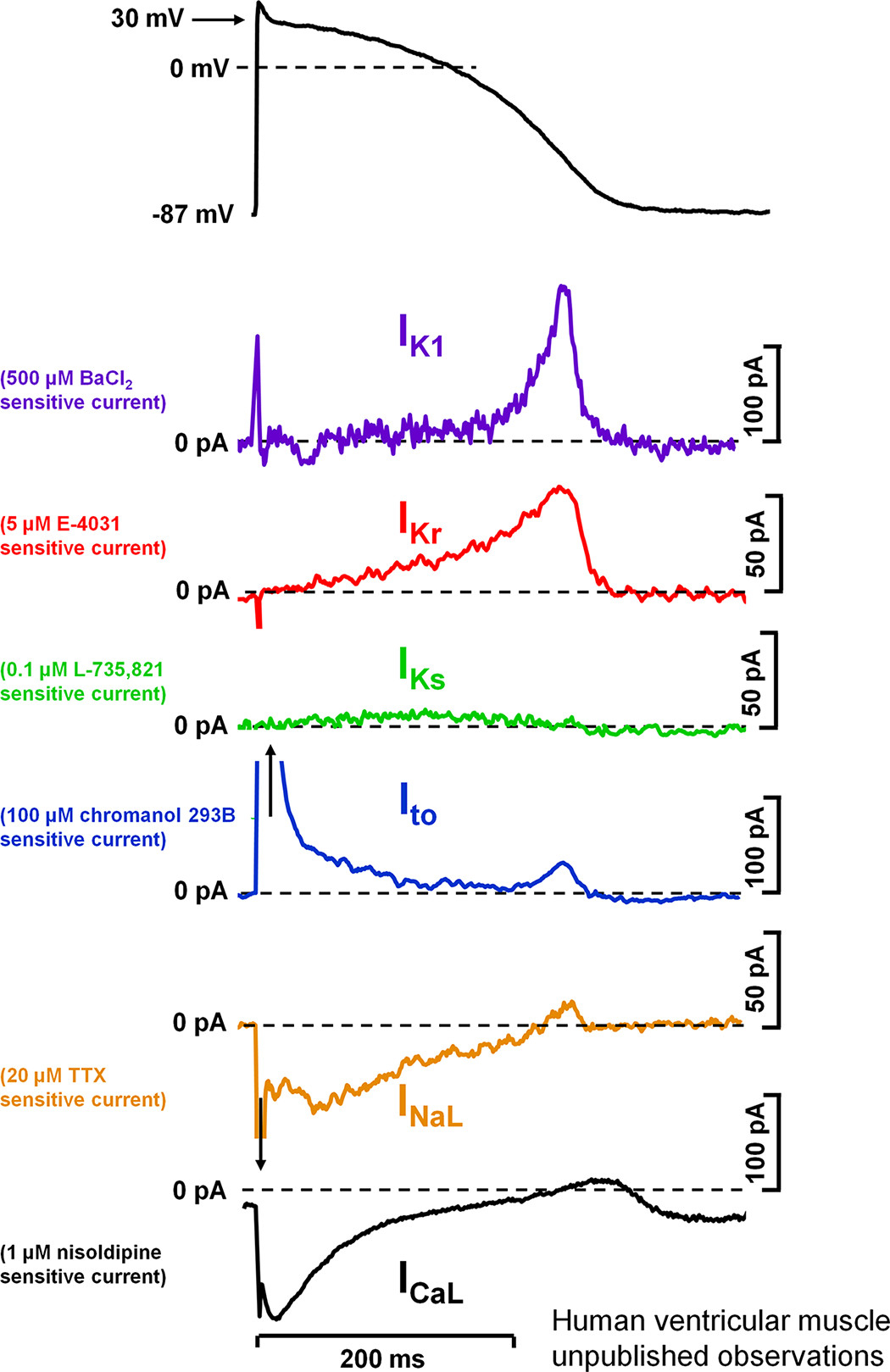
Figure 4 : Action potential and underlying ionic currents recorded from human ventricular myocytes with the patch-clamp technique applying human ventricular action potential as command pulses at 1 Hz stimulation frequency, in the absence of any sympathetic effects. Inward rectifier potassium current (IK1), rapid (IKr) and slow (IKs) components of delayed rectifier potassium current, transient outward current (Ito), and L-type calcium current (ICa,L) were measured as difference current following application of selective channel inhibitors. INaL, late sodium current. Unpublished data from our laboratory at the Department of Pharmacology and Pharmacotherapy,
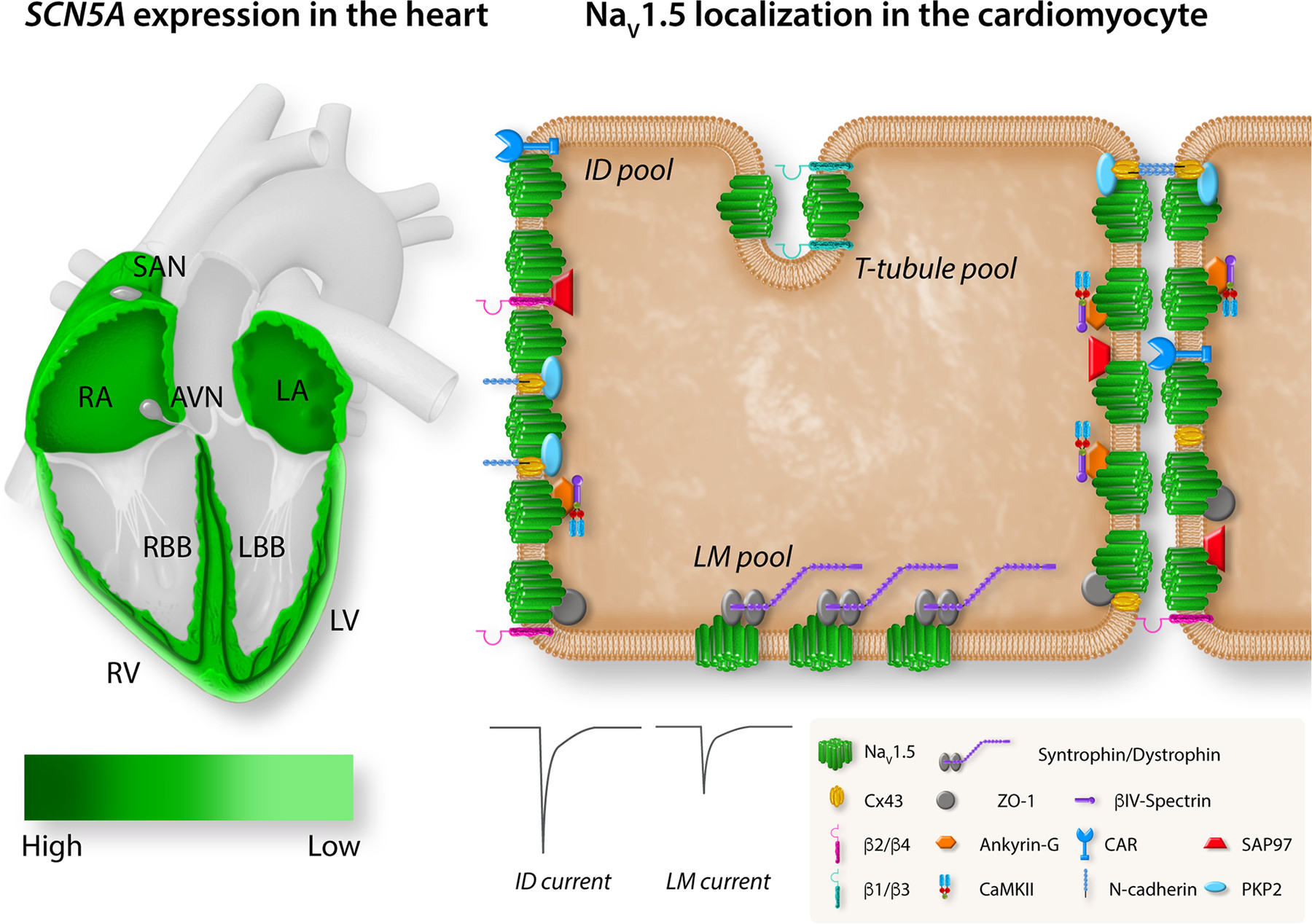
Figure 5 : Regional and subcellular distribution of SCN5A/voltage-gated sodium channel (Nav)1.5 in the heart and cardiomyocytes. Left: the expression levels of SCN5A in different regions of the heart. Expression of SCN5A is highest in the atrioventricular (AV) bundle, His bundle, and right (RBB) and left (LBB) bundle branch (dark green). SCN5A is broadly expressed in right (RA) and left (LA) atria and right (RV) and left (LV) ventricle with an epi/endo gradient in the ventricles. SCN5A is absent from the central sinoatrial node (SAN) and atrioventricular node (AVN). Right: the localization of Nav1.5 with specific regional partner proteins in the microdomains of a cardiomyocyte: intercalated disk (ID), lateral membrane (LM), and T tubules. The sodium current at the ID is larger than the sodium current at the LM. Reproduced from Ref. 94 with permission.
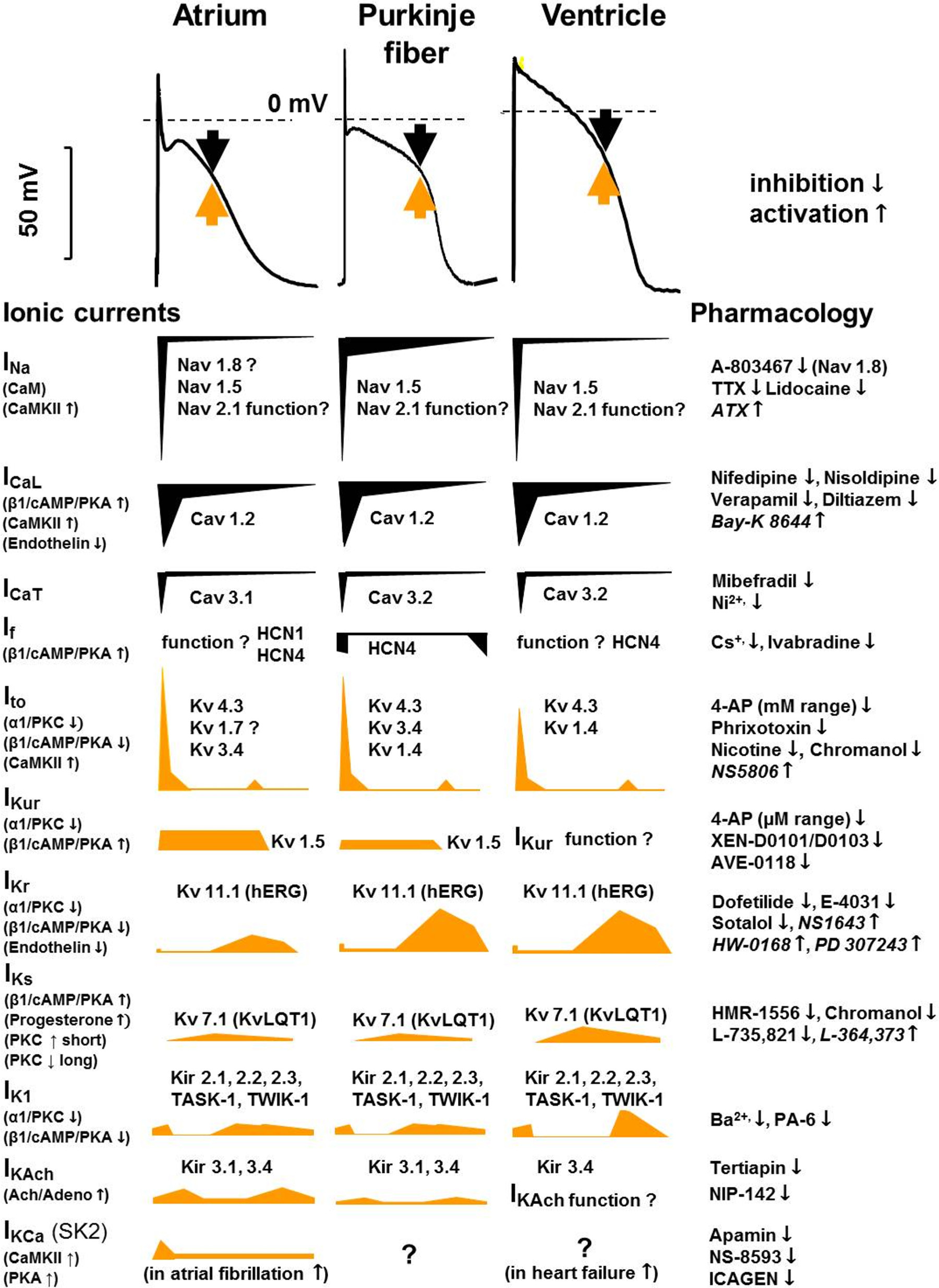
Figure 6 : Tissue-specific (human) cardiac atrial, Purkinje fiber, and ventricular action potentials and the underlying ionic currents in different action potential phases, indicating their pharmacology and modulation. Black arrows indicate inward and yellow arrows indicate outward current. The contributions of different currents to the action potentials are indicated below, with a time course adjusted to the action potential. CaM, calmodulin; CaMKII, Ca2+-calmodulin kinase II; hERG, human ether-à-go-go-related gene; IK1, inward rectifier potassium current; IK,Ach, acetylcholine-activated potassium current; INa, sodium current; ICaL, L-type calcium current; ICaT, T-type calcium current; If, funny/pacemaker current; Ito, transient outward current; IKCa, calcium-activated potassium current; IKr, IKs, and IKur, rapid, slow, and ultrarapid components of delayed rectifier potassium current; Kir, inward rectifier potassium channel; KV, voltage-gated potassium channel; NaV, voltage-gated sodium channel; TASK, Tandem of pore domains in a weak inward rectifying potassium channel (TWIK)-related acid-sensitive potassium channel; TTX, tetrodotoxin.
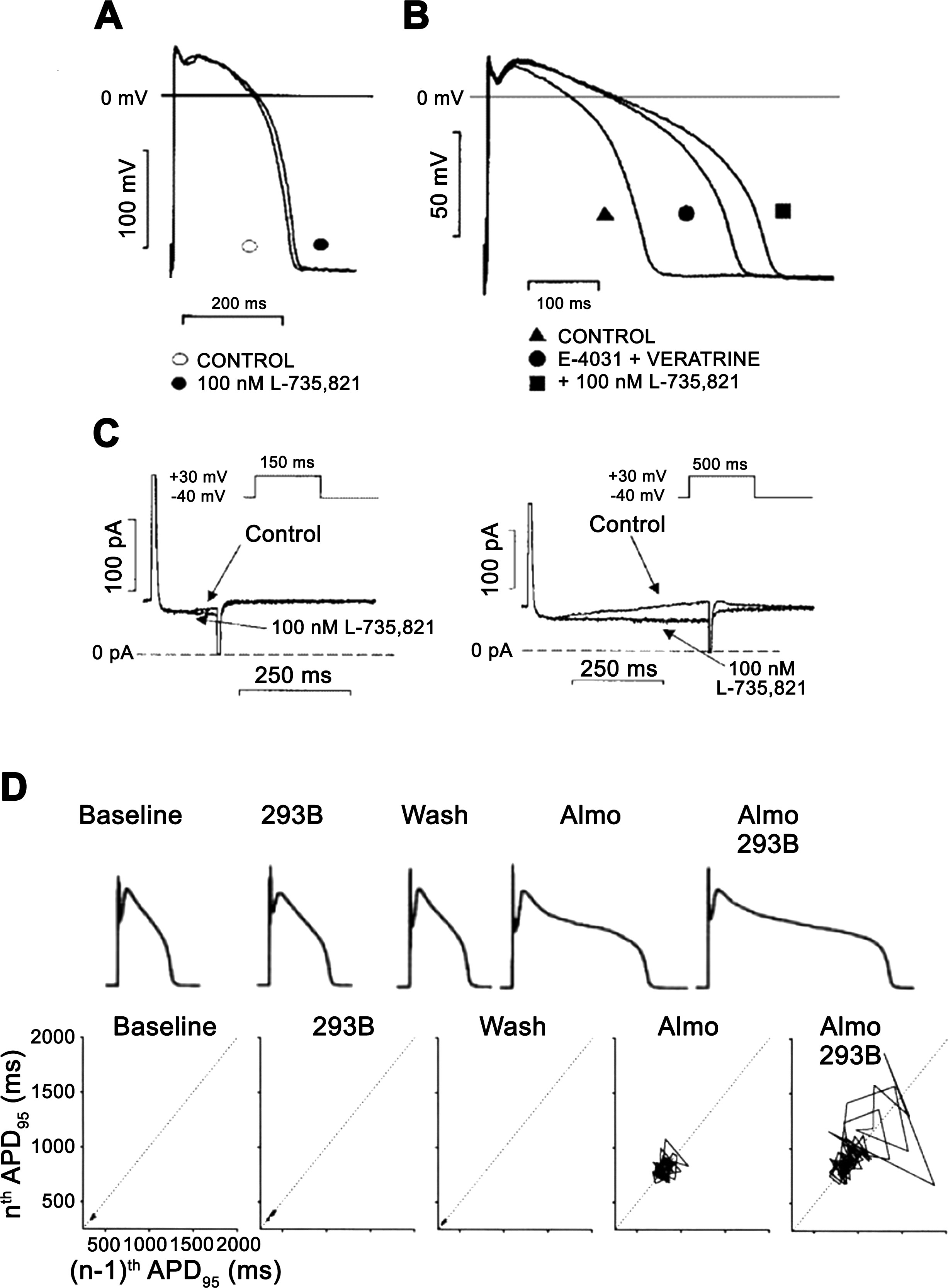
Figure 7 : The role of the slow component of the delayed rectifier potassium current (IKs) in the repolarization in dog ventricular papillary muscle (A–C) and in dog single ventricular myocyte (D). In A, action potential duration (APD) is not, or minimally, changed after full IKs inhibition by 100 nM L-735,821 without external sympathetic stimulation. In B, in the same preparation full IKs inhibition elicited significant prolongation of repolarization in a preparation where the rapid component of the delayed rectifier potassium current (IKr) was inhibited by E-4031 and late sodium current (INaLate) was augmented by veratrine. In C, transmembrane current recordings show that during a short (150 ms) voltage pulse very little IKs develops, but when pulse duration was increased to 500 ms significant IKs developed, explaining the lack and significant changes of APD after IKs inhibition in A and B. (Reproduced from Ref. 228 with permission.) In D, the result of a representative experiment is shown in dog ventricular myocytes. In the baseline (control) situation, small short-term APD variability and normal APD were recorded that did not change significantly after IKs inhibition by chromanol 293B. Additional application of IKr block by almokalant increased both APD and short-term APD variability illustrated by the Poincaré plot, which was substantially further lengthened and increased by additional IKs inhibition, respectively. (Reproduced from Ref. 255 with permission.)
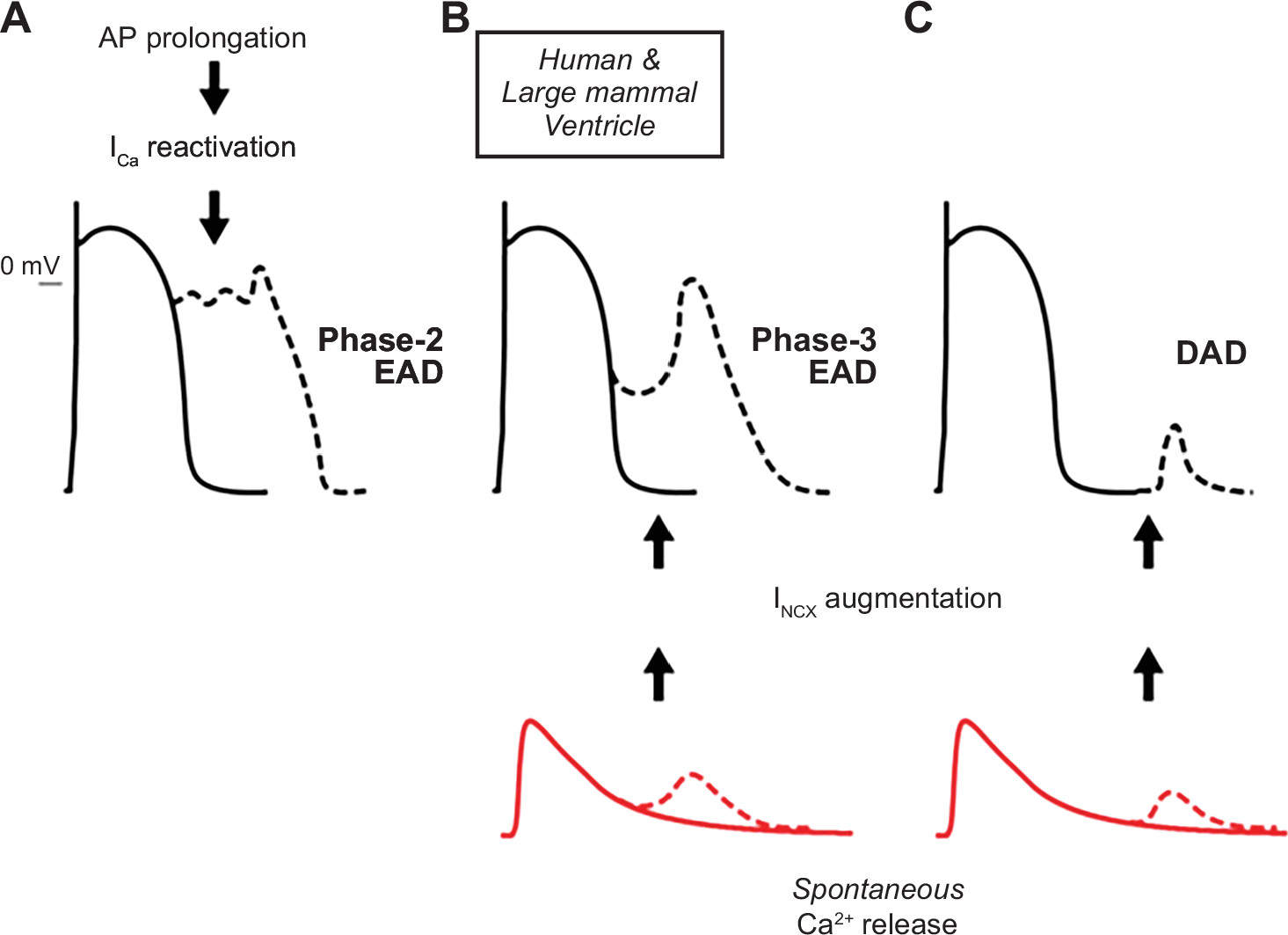
Figure 10 : Mechanisms of afterdepolarization formation in cardiomyocytes. In ventricular myocytes from large mammals, phase 2 early afterdepolarizations (EADs) are associated with L-type Ca2+ current (ICa,L) recovery from inactivation and reactivation during prolonged action potentials (APs) (A). Spontaneous sarcoplasmic reticulum Ca2+ release, which increases Ca2+ extrusion via Na+/Ca2+ exchange (inward current) (INCX), can lead to phase 3 EADs (B) or delayed afterdepolarizations (DADs) (C) when occurring during or after the termination of repolarization, respectively. (Reproduced from Ref. 434 with permission.)
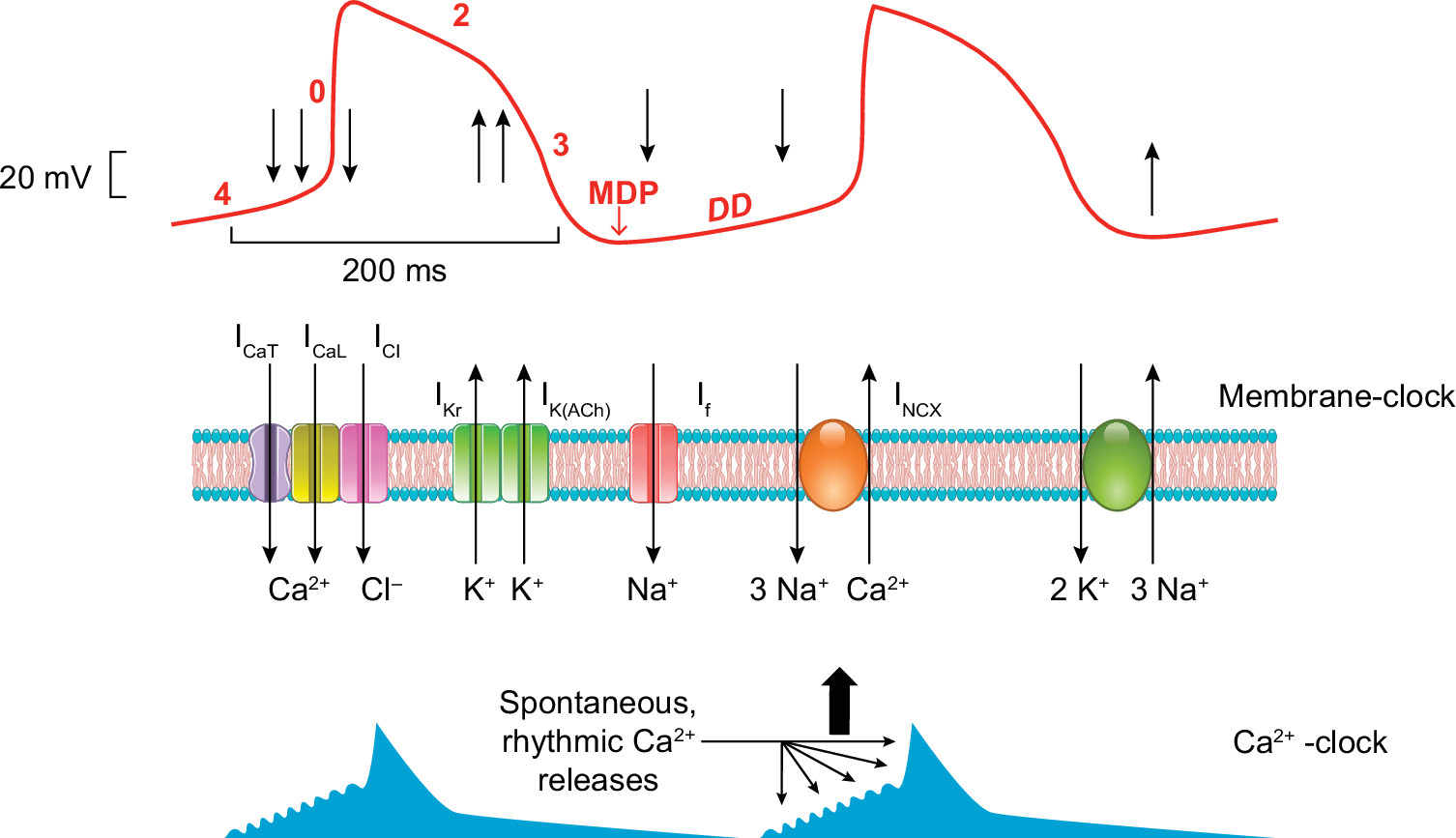
Figure 11 : Hypothetical mechanism for sinus node pacemaking. The figure illustrates a typical sinus node action potential (red trace) and the timing of the membrane and Ca2+ clock components. Upon phase 0, the T (ICa,T)- and L(ICa,L)-type Ca2+ channels (and presumably the Cl− channels, ICl) open, providing inward current (downward arrows), and depolarize the membrane. During repolarization (phase 3) the opening of the delayed rectifier (IKr) and the acetylcholine-dependent (IK,ACh) K+ currents (upward arrows) repolarizes the membrane to reach the maximal diastolic potential (MDP). Upon diastolic depolarization (DD), the cAMP-dependent “funny current” (If) slowly depolarizes the membrane in close cooperation with the sodium-calcium exchanger (INCX) that is governed by spontaneous, rhythmic Ca2+ oscillations from the sarcoplasmic reticulum.
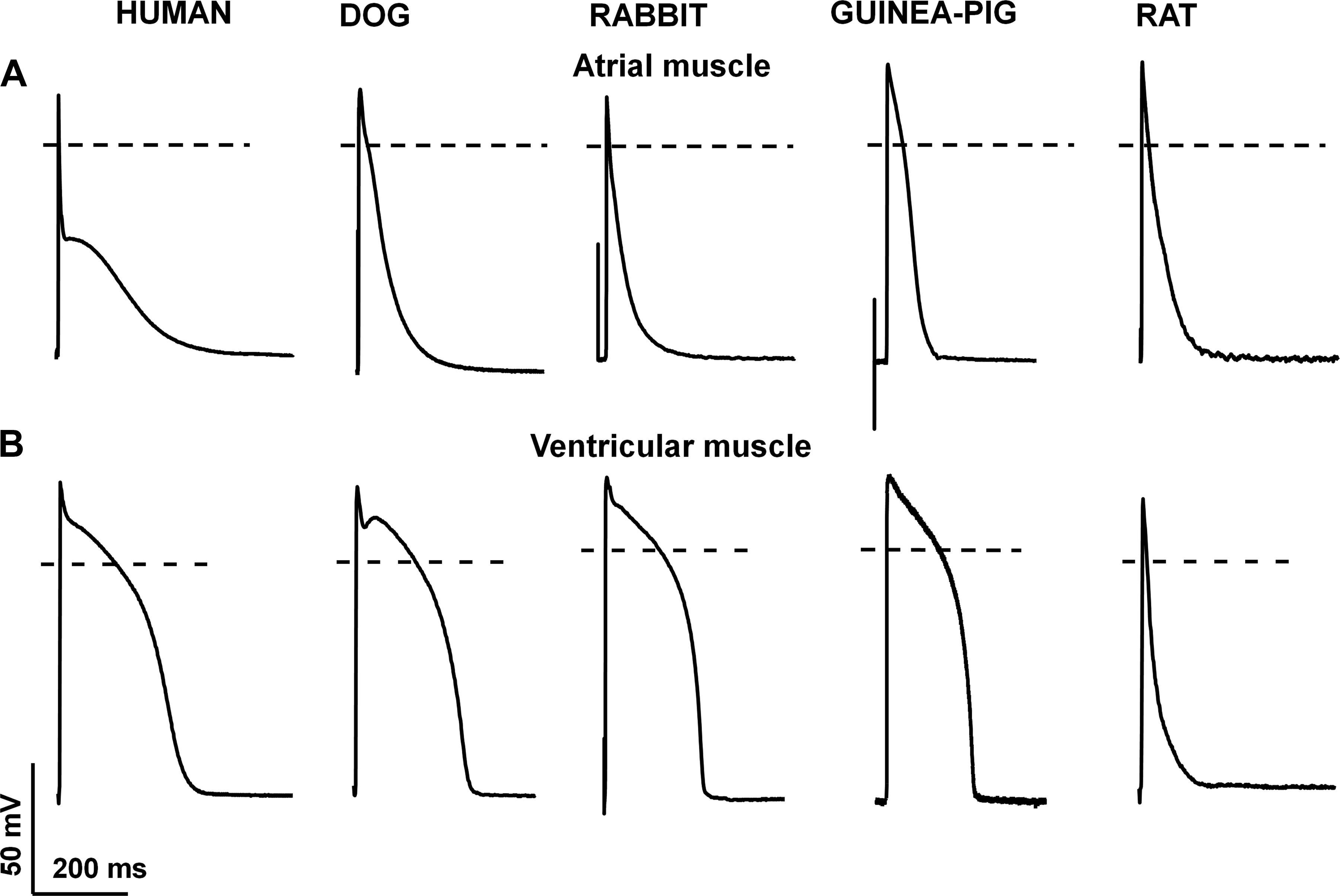
Figure 12 : Species-dependent differences in atrial (A) and ventricular (B) action potential configuration. Original action potential recordings at 1 Hz stimulating frequency from human, dog, rabbit, guinea pig, and rat ventricular muscle preparations recorded by the conventional microelectrode technique. Unpublished data from our laboratory at the Department of Pharmacology and Pharmacotherapy, University of Szeged.
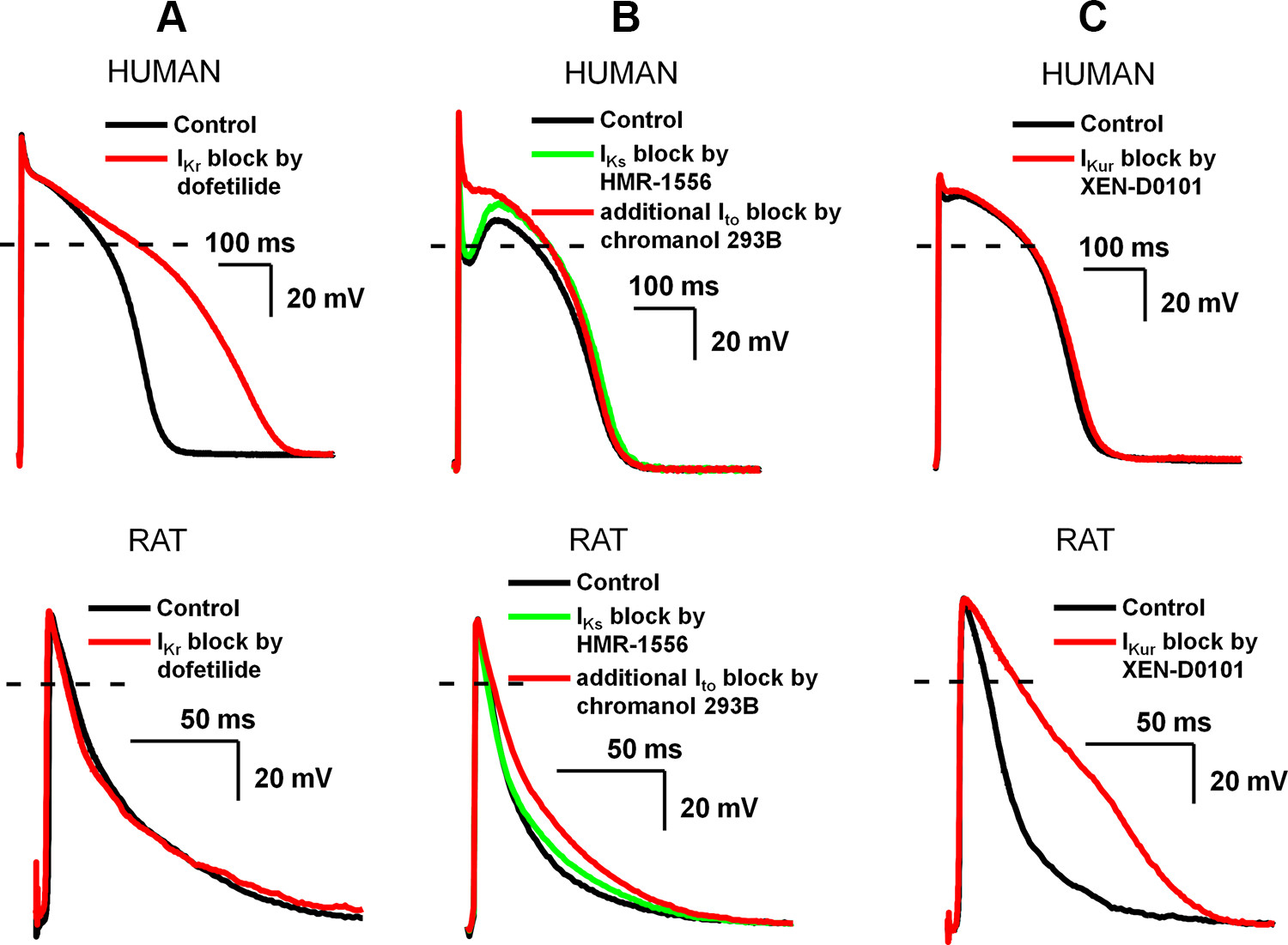
Figure 13 : Distinctly different effects of slow component of delayed rectifier potassium current (IKs), transient outward current (Ito), and ultrarapid component of delayed rectifier potassium current (IKur) inhibition on ventricular repolarization in human (top) and rat (bottom). In these experiments, action potentials were recorded in ventricular papillary muscle with the conventional microelectrode technique at 1 Hz stimulation frequency. Note the extended timescale of the recordings in rat papillary muscle. A: rapid component of delayed rectifier potassium current (IKr) inhibition by 50 nM dofetilide markedly prolonged action potential duration (APD) in human but not rat papillary muscle. B: Ito block by 100 µM chromanol 293B in human eliminated the notch and elevated the plateau potential to positive direction without changing the APD; however, the APD was significantly prolonged in rat. Since chromanol 293B fully blocks IKs in addition to inhibition of Ito at 100 µM, prior IKs block was elicited by 500 nM HMR-1556 to dissect these effects (140). C: IKur inhibition by 3 µM XEN-D0101 (310) does not affect APD in human but markedly prolongs APD in rat ventricular papillary muscle (unpublished experiments from the Department of Pharmacology and Pharmacotherapy, University of Szeged).
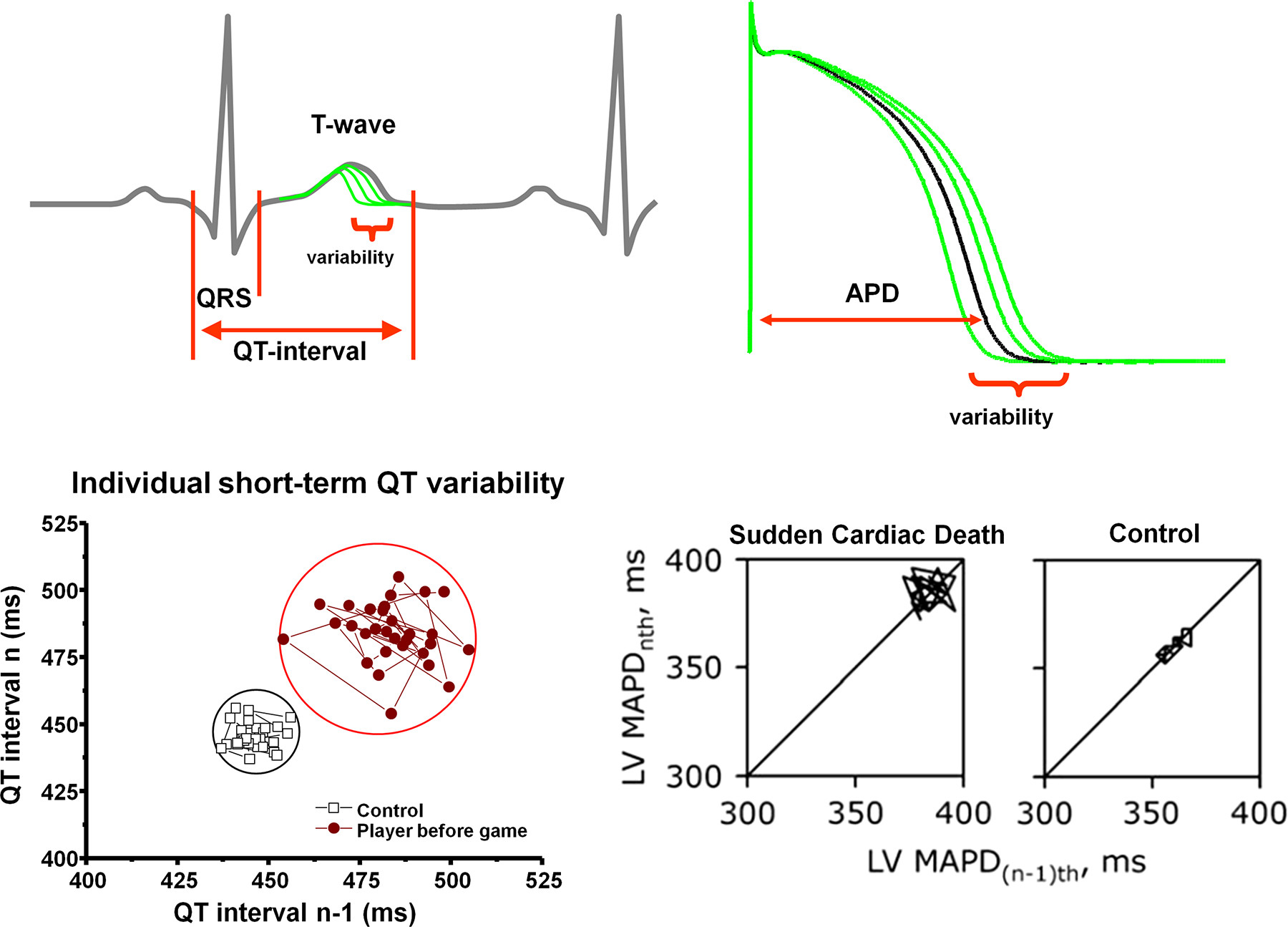
Figure 20 : Short-term temporal variability of repolarization and the risk of sudden cardiac death (SCD). Top left: schematic illustration of QT interval variability on an electrocardiogram trace. Top right: schematic illustration of action potential duration (APD) variability. Bottom left: increased QT interval short-term variability in a representative professional soccer player compared with a control individual, illustrated on a Poincaré plot, where each QT interval n is plotted against its former value, n − 1. Bottom right: individual Poincaré plots illustrating increased short-term variability of left ventricular (LV) monophasic action potential duration (MAPD) in chronic atrioventricular block dogs with SCD compared with control dogs without SCD. (Modified from Ref. 861 with permission.)