Topic 1
The route of entry into the body for SARS-CoV-2 is by attaching to apical membrane proteins of epithelial cells.
The type of epithelial cells that express these proteins will define the organ systems initially involved and influence the progression of disease.
Develop the progression of infection at the initial sites of viral contact and as it continues to other body locations.
A.) Examine the cellular localizations of membrane bound angiotensin-converting-enzyme2 ( ACE2 ) that SARS-CoV-2 binds to via its spike protein , along with the proteases that prime the SARS-CoV-2 spikes for viral internalization ( furin , TMPRSS2 )
ACE2 Localization:
ACE2 is highly expressed on the apical surface of ciliated epithelial cells in the nasal epithelium, making it a key site for initial SARS-CoV-2 infection. Expression diminishes in a gradient from the upper respiratory tract (nasal cavity) to the lower respiratory tract (bronchioles and alveoli).
In the alveoli, type II pneumocytes express ACE2 at lower levels compared to nasal cells. However, these cells are still vulnerable, contributing to severe lung pathologies like acute respiratory distress syndrome (ARDS).
Proteases (TMPRSS2 and Furin):
TMPRSS2: A serine protease that primes the spike protein for membrane fusion. It is expressed in the same regions as ACE2, enabling efficient viral entry.
Furin: Located intracellularly and extracellularly, furin cleaves the spike protein at the S1/S2 junction. This cleavage is critical for the conformational changes required for viral fusion with host cell membranes.
Synergy: The co-expression of ACE2 and these proteases on specific epithelial cells explains the tissue tropism of SARS-CoV-2.
B.) Consider how cellular heterogeneity of infection could selectively compromise specific functions.
Selective Infection:
SARS-CoV-2 selectively infects cells expressing both ACE2 and TMPRSS2. Ciliated airway cells and secretory (club) cells are particularly targeted.
Non-uniform expression of these proteins creates cellular heterogeneity, meaning not all cells in a tissue are equally susceptible. This heterogeneity leads to patchy infection patterns and differential impacts on tissue functions.
Functional Consequences:
Infection of ciliated cells disrupts mucociliary clearance, a defense mechanism of the respiratory tract.
Secretory cells, which produce protective mucus, may show less infection but could still suffer indirect effects, like inflammation or compromised repair mechanisms.
C.) Examine the progression of infection along various sites in the respiratory tract
Initial Infection:
The nasal cavity serves as the primary site of infection due to high ACE2 expression on ciliated cells. This early stage often correlates with mild symptoms like anosmia or rhinorrhea.
Lower Respiratory Involvement:
Aspiration of viral particles leads to infection of lower respiratory structures, including the bronchioles and alveoli. Type II pneumocytes, which produce surfactant, are particularly susceptible.
The gradient of ACE2 and TMPRSS2 expression explains why the upper respiratory tract is more frequently affected but also why severe disease manifests in the lungs.
Clinical Implications:
The viral load increases as the infection progresses down the tract, leading to complications such as pneumonia, ARDS, and in severe cases, multi-organ involvement.
D.) Examine possible mechanisms for loss of smell associated with SARS-CoV-2 infection.
Mechanism:
Supporting cells in the olfactory epithelium express high levels of ACE2. These cells provide structural and metabolic support to sensory neurons.
Infection and inflammation in the olfactory epithelium can disrupt these support functions, leading to temporary anosmia.
Significance:
Loss of smell often occurs early in the disease and may not correlate with severe systemic symptoms. It serves as an important diagnostic clue for early detection of SARS-CoV-2 infection.
E.) Consider the consequences of ACE2 internalization and degradation during SARSCoV-2 infection on the balance of angiotensin dependent signaling at the various angiotensinergic receptors
ACE2 Internalization:
Binding of the SARS-CoV-2 spike protein triggers ACE2 internalization and degradation, reducing its availability on the cell surface.
Angiotensin Signaling Imbalance:
ACE2 normally converts angiotensin II into angiotensin (1–7), which has anti-inflammatory and vasodilatory effects.
Decreased ACE2 leads to an accumulation of angiotensin II, promoting:
Vasoconstriction
Inflammation
Oxidative stress
This imbalance exacerbates lung injury and contributes to systemic complications.
F.) Consider the different physiologic outcomes for apical versus basolateral exit of SARSCoV-2 from epithelial and endothelial cells.
Apical Exit:
Virus released from the apical side of epithelial cells spreads along the mucosal surfaces, facilitating respiratory transmission.
This mechanism primarily drives person-to-person transmission.
Basolateral Exit:
Basolateral release allows access to the underlying vasculature or lymphatics, promoting systemic dissemination.
This route could lead to viremia and infection of extrapulmonary sites, contributing to multi-organ damage.
G.) Consider what features of the placenta may protect the fetus from direct SARS-CoV-2 infection.
The placenta has low ACE2 expression, limiting the potential for direct viral entry.
Physical barriers, like the syncytiotrophoblast layer, and strong innate immune responses protect the fetus.
However, inflammatory cytokines associated with severe maternal infection may pose indirect risks to fetal development.
H.) Examine how vaccines mimic viral infection to generate an immune response
Mechanism of Action:
Vaccines deliver the spike protein (or its genetic blueprint) to the immune system, mimicking a natural infection.
Humoral Response: B cells produce neutralizing antibodies targeting the spike protein, preventing viral entry into cells.
Cellular Response: T cells recognize and destroy infected cells, curbing viral replication.
Immune Memory:
Vaccines also generate memory B and T cells, providing long-term protection and quicker responses upon future exposures.
Topic 2
Respiratory infection by SARS-CoV-2 can impair gas exchange and require intense clinical intervention.
Develop a physiologic frame-work for the consequences of prolonged infection in the respiratory tract.
A.) Compare possible mechanisms that would lower blood O2 saturation during CoViD-19 progression.
ENaC Dysfunction :
Alveolar type II ( AT2 ) cells are essential for producing surfactant and reabsorbing alveolar fluid through epithelial sodium channels ( ENaC )
Infection inhibits ENaC activity , leading to fluid retention in the alveoli , alveolar flooding ( edema ) , and impaired gas exchange
also reduces alveolar fluid clearance ( AFC ) and disrupting mucociliary clearance ( MCC )
Endothelialitis / Cytokine Storm :
the virus can infect endothelial cells , causing inflammation
this increases capillary permeability , contributing to alveolar flooding and promoting thrombotic complications like microvascular clots that block pulmonary circulation , further impairing oxygenation
Impaired Mucociliary Clearance ( MCC ) :
Infected ciliated cells in the respiratory tract disrupt mucociliary clearance , slowing the removal of pathogens and increasing the risk of distal airway infection and obstruction.
This exacerbates airway inflammation and impairs ventilation
critically depend on an amiloride-sensitive ENaC-dependent sodium transport expressed along the entire respiratory tract.
Acute Respiratory Distress Syndrome ( ARDS ) :
In advanced stages , the cumulative effects of alveolar flooding , inflammatory exudates , and reduced surfactant lead to ARDS
This condition severely reduces lung compliance and oxygen diffusion , drastically lowering blood O2 saturation
Impaired Vascular Response :
Dysfunctional endothelial signaling can disrupt vasodilation mechanisms , reducing perfusion to well-ventilated lung areas ( ventilation-perfusion mismatch ).
Additionally, hypoxia-induced vasodilation inappropriately perfuses flooded alveoli
Ventilation-Perfusion ( V / Q ) Mismatch :
Mechanism : Endothelial dysfunction and hypoxic vasodilation prevent redirection of blood flow from non-functional alveoli, worsening oxygenation.
Impact : Blood flows to poorly ventilated areas, further reducing overall gas exchange efficiency.
Reduced Oxygen Transport :
Coagulopathy and systemic inflammation can impair oxygen delivery by disrupting vascular integrity and causing hypoxic damage to other organs, including the heart
Hypoxic Vasoconstriction Failure :
Local vasodilation due to inflammatory mediators like nitric oxide prevents redirection of blood away from non-functional alveoli , reducing overall oxygenation.
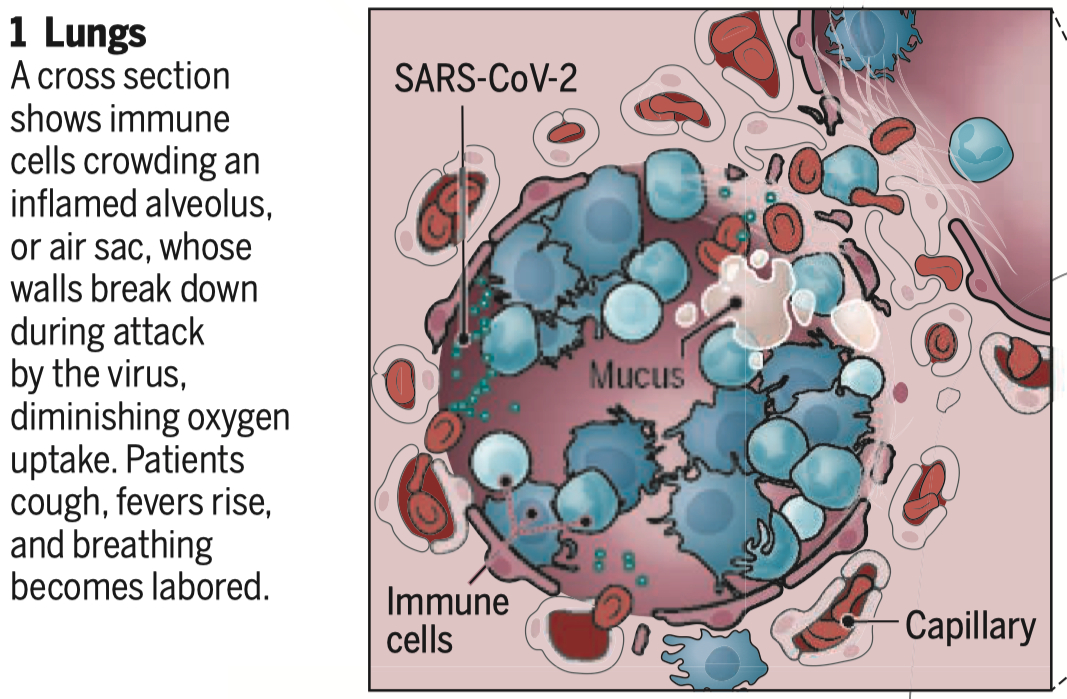
B.) Examine the alterations in gas exchange due to increases in the apical fluid layer of alveolar epithelial cells during inflammatory transudation.
Alveoli rely on alveolar type 2 ( AT2 ) cells to maintain proper fluid balance via sodium transport , largely mediated by the epithelial sodium channel ( ENaC )
ENaC function supports alveolar fluid clearance ( AFC ) , essential for gas exchange.
Inflammation generated cytokines ( e.g., TNF-α , IL-6 ) cause inhibition of ENaC
Inhibiting ENaC ➡️ increased apical fluid in alveoli ( edema )
Edema disrupts the air-liquid interface crucial for efficient gas exchange
Eventually results in hypoxemia and acute respiratory distress syndrome ( ARDS )
Mechanisms of ENaC Inhibition :
Competitive binding : Viral proteins or inflammatory mediators can compete for proteolytic enzymes like furin , preventing ENaC activation.
Gene transcription block: Viruses may suppress the transcription of ENaC subunit genes, further reducing sodium transport efficiency.
Clinical Implications :
Increased apical fluid due to transudation exacerbates ARDS and contributes to the hypoxia seen in severe cases.
Targeting ENaC preservation or enhancing its activity could be therapeutic strategies to mitigate gas exchange impairment during inflammatory conditions.
C.) Consider the influence of increased endothelial permeability of pulmonary capillaries in response to the inflammation instigated by a prolonged immune cell release of pro-inflammatory cytokines.
Endothelial Permeability :
Pro-inflammatory cytokines like TNF-α and IL-1β disrupt the tight junctions between endothelial cells, leading to increased capillary leakage.
This leakage allows proteins and fluids to accumulate in the alveolar spaces, resulting in pulmonary edema.
Cytokine Storm :
Excessive cytokine release intensifies oxidative stress and endothelial dysfunction.
This exacerbates vascular barrier disruption, impairing alveolar fluid clearance and leading to hypoxemia and ARDS.
Alveolar Fluid Dynamics :
Normally, AT2 cells reabsorb alveolar fluid via ENaC
Cytokines inhibit ENaC activity, worsening pulmonary edema and reducing oxygen exchange.
This leads to ARDS , characterized by severe hypoxemia and impaired gas exchange
D.) Examine the outcomes of pulmonary clot formation due to increased coagulation events stemming from a hyper-inflammatory condition.
Endothelial Dysfunction :
Inflammatory cytokines , such as IL-1 and TNF, activate the endothelium , leading to :
the expression of tissue factor, a potent procoagulant
and downregulation of anticoagulant mechanisms like thrombomodulin.
This creates a pro-thrombotic environment within pulmonary vessels
Microvascular Thrombosis :
Activation of coagulation cascades , compounded by reduced fibrinolysis due to elevated plasminogen activator inhibitor-1 (PAI-1) ,
results in the formation of clots in the pulmonary microcirculation.
This occludes blood flow , leading to hypoxia and contributing to ARDS
Systemic Effects :
Widespread thrombus formation contributes to systemic hypoxia, multi-organ failure, and ARDS progression
Clinical Outcomes :
Impaired perfusion from clots disrupts ventilation-perfusion matching, worsening oxygenation.
Therapies targeting coagulation cascades (e.g., anticoagulants) are critical in managing severe cases.
E.) Consider the alterations of airway dead space during mechanical ventilation.
Impact of Mechanical Ventilation :
Intubation increases physiologic dead space by overdistending alveoli, reducing ventilation efficiency.
Positive pressure can exacerbate ventilation-perfusion mismatches.
Effects on Mucociliary Clearance :
SARS-CoV-2 disrupts ciliated cell function, impairing mucociliary clearance (MCC) and increasing mucus retention.
This further increases airway obstruction and dead space.
Alveolar Dynamics :
Edema caused by cytokine-mediated ENaC inhibition reduces effective alveolar ventilation.
Vascular and endothelial dysfunction adds to the mismatch by impairing blood flow to ventilated areas.
Key Considerations :
Optimizing ventilator settings to minimize alveolar overdistension and dead space is essential.
Addressing underlying inflammation and edema is crucial for restoring ventilation efficiency.
F.) Consider the respiratory changes associated with wearing a tight-fitting mask ( either N-95 or 2-layer cloth homemade ) , including flow resistance and humidification as well as sensory feedback leading to feelings of discomfort.
Flow Resistance:
What: Tight-fitting masks like N95s have a dense filtration layer that increases airflow resistance.
Why: This increases the effort required to inhale, leading to higher work of breathing, particularly during exertion.
Impact: Over time, individuals may experience fatigue or mild hypercapnia (elevated CO2) due to restricted exhalation and rebreathing of trapped exhaled air.
Humidification:
What: The mask traps moisture from exhaled breath, increasing localized humidity.
Why: Reduced air exchange within the mask confines moisture.
Impact: This can affect mucosal hydration and potentially lead to discomfort, but it may also benefit airway health by reducing dryness.
Sensory Feedback and Discomfort:
What: Pressure points, humidity, and airflow resistance provide tactile and sensory signals.
Why: These signals are interpreted by the brain as discomfort, which may lead to heightened awareness of breathing and possibly a subjective sense of suffocation, even if oxygen exchange is unaffected.
Impact: This can result in reduced mask compliance or increased anxiety in some individuals.
Effects on Gas Exchange:
What: Masks may create a small dead space for exhaled air, leading to a minimal increase in inhaled CO2 levels.
Why: Rebreathing exhaled air trapped in the mask pocket contributes to this effect.
Impact: In healthy individuals, this is usually negligible but may cause mild hypercapnia in people with compromised respiratory function.
Increased Resistance: Tight masks like N-95 respirators increase airflow resistance, requiring greater effort to breathe.
Humidification: Masks trap exhaled moisture, increasing airway hydration and potentially altering sensory feedback.
Discomfort: Sensory input from mask wear may increase perceived respiratory effort or anxiety, especially during prolonged use
Topic 3
The progression of CoViD-19 can lead to alterations in control of blood flow due to the binding of SARS-CoV-2 to ACE2
Develop a physiologic frame-work for the consequences of renal SARS-CoV-2 infection.
A.) Examine the possibility that SARS-CoV-2 is filtered at the glomerulus and enters renal tubular fluid. Consider the consequences of infection for podocytes on glomerular filtration, including selectivity and total permeability ( filtration coefficient )
SARS-CoV-2 binds to ACE2 on podocytes and tubular cells, entering these cells and causing damage.
Podocyte injury disrupts the glomerular filtration barrier, leading to:
Proteinuria due to loss of selectivity.
Decreased filtration coefficient (Kf) from reduced filtration surface area.
Chronic podocyte damage can result in glomerulosclerosis and progression to kidney disease.
B.) Examine how blocking angiotensin receptors would alter angiotensin signaling during CoViD-19 progression.
Blocks angiotensin-II (ANG-II) action, reducing vasoconstriction and inflammation.
Promotes protective ACE2/ANG-(1–7)/Mas axis effects, such as vasodilation and anti-inflammation.
Reduces hypertension and vascular permeability, alleviating fluid retention and pulmonary complications.
C.) Consider the renal consequences of contraction of systemic arterioles by angiotensin-II. Remember that angiotensin-II also stimulates aldosterone release from adrenal glands. Include changes in extracellular fluid volume over the course of several days, with ideas concerning distal Na + absorption as well as the release of antidiuretic hormone.
Systemic arteriolar contraction raises blood pressure and total peripheral resistance.
Aldosterone release increases distal sodium reabsorption, expanding extracellular fluid (ECF) volume over days.
ANG-II stimulates ADH secretion, enhancing water retention and further expanding ECF.
Prolonged fluid retention exacerbates risks like hypertension and edema.
D.) Consider how CoViD-19 progression could lead to acute kidney failure
Direct infection of renal cells via ACE2 causes tubular necrosis and podocyte injury.
Cytokine storms induce systemic inflammation and vascular permeability, impairing renal perfusion.
Hypercoagulability leads to microthrombosis, reducing effective glomerular filtration.
Dysregulated RAS exacerbates renal ischemia and perfusion deficits.
E.) Consider the possible mechanisms leading to the observed increase in plasma creatinine and urea concentration for some CoViD-19 patients.
Reduced glomerular filtration rate (GFR) decreases clearance of creatinine and urea.
Heightened protein catabolism from cytokine storms increases urea production.
Hypoperfusion and ischemia impair renal clearance of nitrogenous waste products.
F.) Compare possible mechanisms for simultaneously elevated plasma creatinine and elevated albumin in the urine.
Podocyte injury causes glomerular filtration barrier leakage, leading to albuminuria.
Tubular dysfunction reduces reabsorption efficiency, worsening proteinuria and creatinine retention.
Systemic inflammation exacerbates filtration and reabsorption impairments.
G.) Examine the consequences of maternal SARS-CoV-2 infection on placental blood perfusion.
ACE2 dysregulation decreases ANG-(1–7), leading to reduced placental vasodilation and perfusion.
Endothelial dysfunction and microthrombosis compromise oxygen and nutrient delivery to the fetus.
Elevated ANG-II levels heighten pre-eclampsia risk through systemic vasoconstriction.