Many of the RNA molecules in bacteria and virtually all RNA molecules in eukaryotes are processed to some degree after synthesis. Processing can include addition or deletion of nucleotide sequences as well as chemical modification of RNA nucleotides. All of these events can be used to control the posttranscriptional fate of the RNA in the cell. As a result, many mature RNAs are not exact copies of the DNA genes from which they were transcribed. Some of the most interesting molecular events in RNA metabolism occur during posttranscriptional processing. Intriguingly, several of the enzymes that catalyze these reactions have active sites composed of RNA rather than protein. The discovery of these catalytic RNAs, or ribozymes, has brought a revolution in thinking about RNA function and about the origin of life, as we will discuss in Section 26.4.
A newly synthesized RNA molecule is called a primary or precursor transcript. Perhaps the most extensive processing of primary transcripts occurs in eukaryotic precursor mRNAs (pre-mRNAs) and in the tRNAs of both bacteria and eukaryotes. However, many ncRNAs are also processed.
The precursor transcript for a eukaryotic mRNA typically contains sequences encompassing one gene, although the sequences encoding the polypeptide may not be contiguous. Noncoding tracts that break up the coding region of the transcript are called introns, and the coding segments are called exons (see the discussion of introns and exons in DNA in Chapter 24). In a process called RNA splicing, the introns are removed from the pre-mRNA, and the exons are spliced together to form a continuous sequence that specifies a functional polypeptide. Virtually all human genes contain introns, the average being eight introns per gene. Eukaryotic mRNAs are also modified at each end. A modified nucleotide structure called a cap is added at the end. The end is cleaved, and 80 to 250 A residues are added to create a poly(A) “tail.” The sometimes elaborate protein complexes that carry out capping, splicing, and polyadenylation mRNA-processing reactions do not operate independently. They are organized in association with each other and with the phosphorylated CTD of Pol II; each complex affects the function of the others, as outlined in Figure 26-12.
FIGURE 26-12 Formation of the primary transcript and its processing during maturation of mRNA in a eukaryotic cell. (a) Nuclear RNA processing includes addition of a cap, removal of noncoding intron sequences, transcript cleavage, and polyadenylation. These processes predominantly occur cotranscriptionally and are coupled with transcript elongation. The Pol II CTD plays a critical role in coordinating transcription and processing. (b) This electron micrograph shows a chromosome isolated from a Drosophila embryo during gene expression. An unidentified gene is being transcribed by RNA Pol II, and the nascent transcripts can be observed emerging from the DNA. The RNA transcripts are shorter at the end of the gene and longer at the end, consistent with the directionality of transcription. Splicing of this gene occurs cotranscriptionally by the spliceosome and can be observed by shortening of the RNA once a long intron has been removed and by the presence of lariat introns. The transcripts remain attached to the DNA until cleavage occurs at the completion of transcription.
Proteins involved in mRNA transport to the cytoplasm are also associated with the mRNA in the nucleus, and the processing of the transcript is coupled to its transport. In effect, a eukaryotic mRNA, as it is synthesized, is ensconced in an elaborate and dynamic supramolecular messenger ribonucleoprotein (mRNP) complex comprising dozens of proteins. The composition of the mRNP changes as the transcript is processed, transported to the cytoplasm, and delivered to the ribosome for translation. Associated proteins can dramatically modulate the cellular destination, function, and fate of an mRNA.
In addition to splicing and and end modification, individual purine and pyrimidine nucleotides within primary transcripts can undergo chemical modification. Many eukaryotic mRNAs contain modified nucleotides that affect their interactions with RNA-binding proteins and regulate gene expression; however, RNA modification has been best characterized in primary tRNA transcript processing. Many bases and sugars in tRNAs are modified in both bacteria and eukaryotes, including with unusual bases not found in other nucleic acids (see Fig. 26-22). Many ncRNAs also undergo elaborate processing, often involving the removal of segments from one or both ends.
The ultimate fate of any RNA is its complete and regulated degradation. The rate of turnover of RNAs plays a critical role in determining their steady-state levels and the rate at which cells can shut down expression of a gene when its product is no longer needed. During the development of multicellular organisms, for example, certain proteins must be expressed at one stage only, and the mRNA encoding such a protein must be made and destroyed at the appropriate times.
Eukaryotic mRNAs Are Capped at the End
Most eukaryotic mRNAs have a cap, a residue of 7-methylguanosine linked to the -terminal residue of the mRNA through an unusual ,-triphosphate linkage (Fig. 26-13). The cap helps protect mRNA from ribonucleases. It also binds to specific cap-binding complexes of proteins and participates in binding of the mRNA to the ribosome to initiate translation (Chapter 27).
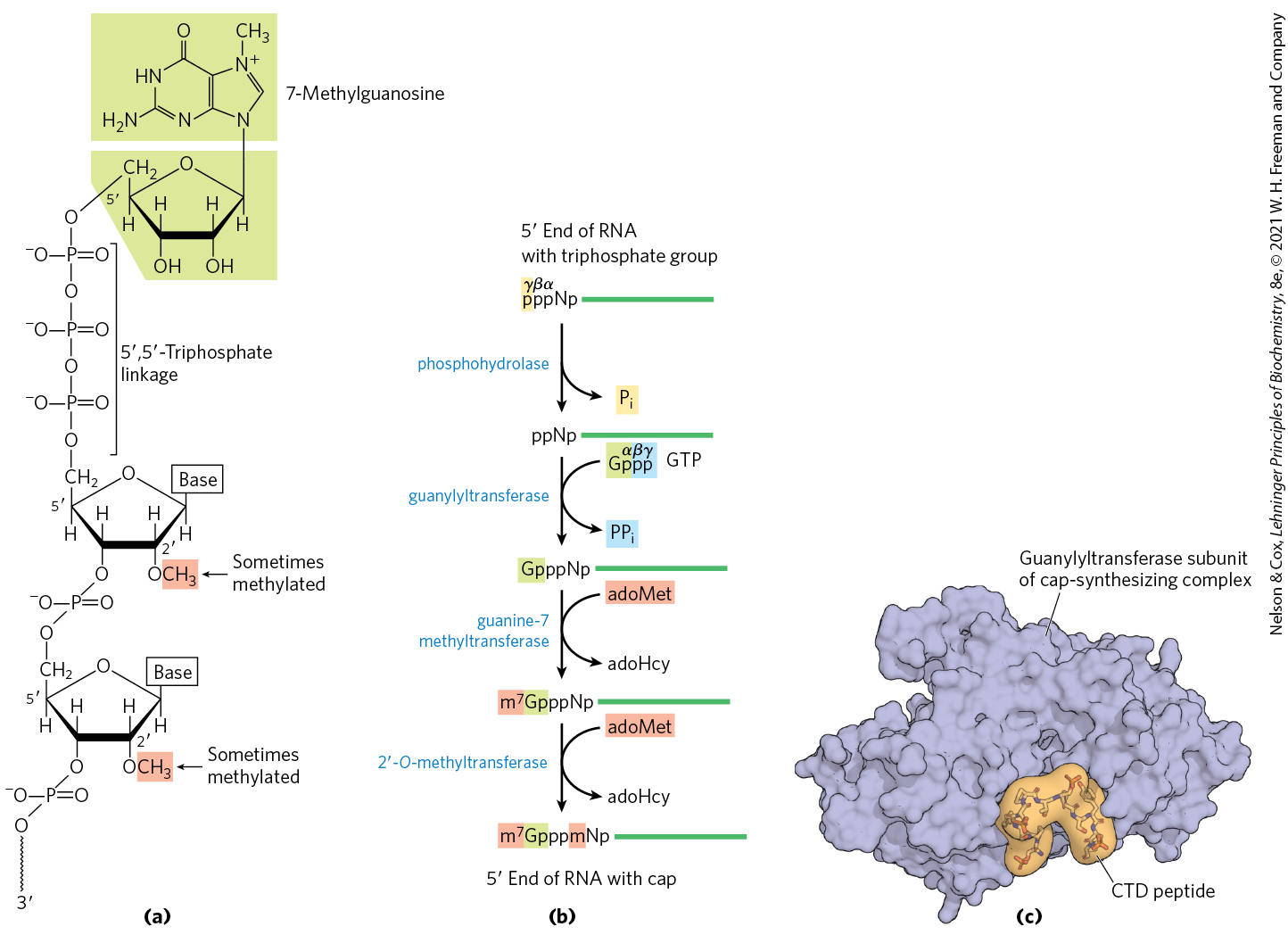
FIGURE 26-13 The cap of mRNA. (a) 7-Methylguanosine () is joined to the end of almost all eukaryotic mRNAs in an unusual ,-triphosphate linkage. Methyl groups (shaded) are often found at the position of the first and second nucleotides in vertebrate cells. (b) Generation of the cap requires four separate steps (adoHcy is S-adenosylhomocysteine). (c) Synthesis of the cap is carried out by enzymes tethered to the CTD of Pol II. Shown here is the structure of the guanylyltransferase subunit of the mouse capping enzyme in complex with a peptide mimicking the Pol II CTD repeat sequence (YSPTSPS). The guanylyltransferase specifically recognizes the first residue (Tyr) and the phosphorylated form of the fifth residue (Ser). [(c) Data from PDB ID 3RTX, A. Ghosh et al., Mol. Cell 43:299, 2011.]
The cap is formed by condensation of a molecule of GTP with the triphosphate at the end of the transcript. The guanine is subsequently methylated at N-7, and additional methyl groups are often added at the hydroxyls of the first and second nucleotides adjacent to the cap (Fig. 26-13a). The methyl groups are derived from S-adenosylmethionine. All these reactions (Fig. 26-12b) occur very early in transcription, after the first 20 to 30 nucleotides of the transcript have been added. All four of the enzymes in the cap-synthesizing complex, and through them the end of the transcript itself, are associated with the RNA polymerase II CTD (Fig. 26-13c) until the cap is synthesized. The capped end is then released from the cap-synthesizing complex and bound by the nuclear cap-binding complex, which facilitates both splicing and nuclear export of the RNA.
The cap does not provide permanent protection of the transcript. Eukaryotes also contain cellular decapping enzymes, which are important for RNA regulation. Cap removal allows RNAs to be degraded by exonucleases that hydrolyze the RNA in the direction. Some viruses have also evolved elaborate mechanisms for removing the cap from host mRNAs. The influenza virus needs no specialized enzymes for the synthesis of caps on its viral RNAs; instead, it borrows these structures from host-cell transcripts in a process termed “cap-snatching.” A capped host transcript is bound by the viral RNA polymerase and cleaved by an endonuclease. The influenza RNA polymerase can then use the resulting capped oligonucleotide to prime viral RNA synthesis.
Both Introns and Exons Are Transcribed from DNA into RNA
In bacteria, the mRNA used for translation is generally a direct copy of the DNA gene sequence, continuing along the DNA template without interruption until the information needed to specify the polypeptide is complete. However, the notion that all genes are continuous was disproved in 1977 when Phillip Sharp and Richard Roberts independently discovered that many genes for polypeptides in eukaryotes are interrupted by noncoding sequences (introns).
The vast majority of genes in vertebrates contain introns; among the few exceptions are those that encode histones. The occurrence of introns in other eukaryotes varies. Many genes of the yeast Saccharomyces cerevisiae lack introns, but introns are more common in some other yeast species. Introns are also found in a few bacterial and archaeal genes. Introns in DNA are transcribed along with the rest of the gene by RNA polymerases. The introns in the primary RNA transcript are then spliced, and the exons are joined to form a mature, functional RNA. In eukaryotic mRNAs, most exons are less than 1,000 nucleotides long, with many in the 100 to 200 nucleotide size range, encoding stretches of 30 to 60 amino acids within a longer polypeptide. Introns vary in size from 50 to more than 700,000 nucleotides, with a median length of about 1,800. Genes of higher eukaryotes, including humans, typically have much more DNA devoted to introns than to exons. For example, the human dystrophin gene encodes a pre-mRNA more than 2 million nucleotides long. However, the final mRNA is 14,000 nucleotides long, indicating that more than 99% of the transcribed RNA is found within introns and removed by splicing. Deficiencies in dystrophin expression can lead to muscular dystrophies. The ~20,000 genes of the human genome include more than 200,000 introns.
RNA Catalyzes the Splicing of Introns
There are four classes of introns (Table 26-3). The first two, the group I and group II introns, differ in the details of their splicing mechanisms but share one surprising characteristic: they are self-splicing — no proteins are needed to carry out catalysis. The introns found in the nuclear-encoded genes of eukaryotes comprise the third class. These pre-mRNA introns are removed by a large RNP called the spliceosome. Although the spliceosome requires dozens of proteins for its function, its active site includes RNA. The final class of introns requires protein enzymes for their removal. These introns are found in some tRNAs as well as certain mRNAs, such as that encoding the Xbox binding protein 1, Xbp1. Protein-mediated splicing of the XBP1 transcript regulates the cellular response to unfolded proteins that occurs under conditions of endoplasmic reticulum stress in human cells. The mechanisms of tRNA and XBP1 mRNA splicing are similar.
TABLE 26-3 Mechanisms of RNA Splicing
Mechanism
Components
Features
Cellular locations
Group I Intron
Catalytic RNA
Self-splicing using a guanine-derived cofactor
Found in nuclear, mitochondrial, and chloroplast genes that encode mRNAs, rRNAs, or tRNAs. Can be found in bacteria.
Group II Intron
Catalytic RNA; maturase and reverse transcriptase proteins
Self-splicing using a nucleophile within the intron to form a lariat
Primarily found in mitochondrial and chloroplast genes of fungi, algae, and plants. Can be found in bacteria.
Spliceosome
Catalytic snRNAs; dozens of protein splicing factors
Requires a large RNP for processing using a nucleophile within the intron to form a lariat
Found in nuclear genes of eukaryotes. Capable of alternative splicing to create multiple products from a given transcript.
Protein-catalyzed
Protein enzymes
Uses a splicing endonuclease and ligase
Found in tRNAs and a few mRNAs.
Group I introns are found in some nuclear, mitochondrial, and chloroplast genes that code for rRNAs, mRNAs, and tRNAs. Group II introns are generally found in the primary transcripts of mitochondrial or chloroplast mRNAs in fungi, algae, and plants. Group I and group II introns are also among the rare examples of introns in bacteria. The splicing mechanisms in both groups involve two transesterification reaction steps (Fig. 26-14), in which a ribose - or -hydroxyl group makes a nucleophilic attack on a phosphorus, and a new phosphodiester bond is formed at the expense of the old, maintaining the balance of energy. These reactions are very similar to the DNA breaking and rejoining reactions promoted by topoisomerases (see Fig. 24-18) and site-specific recombinases (see Fig. 25-37).
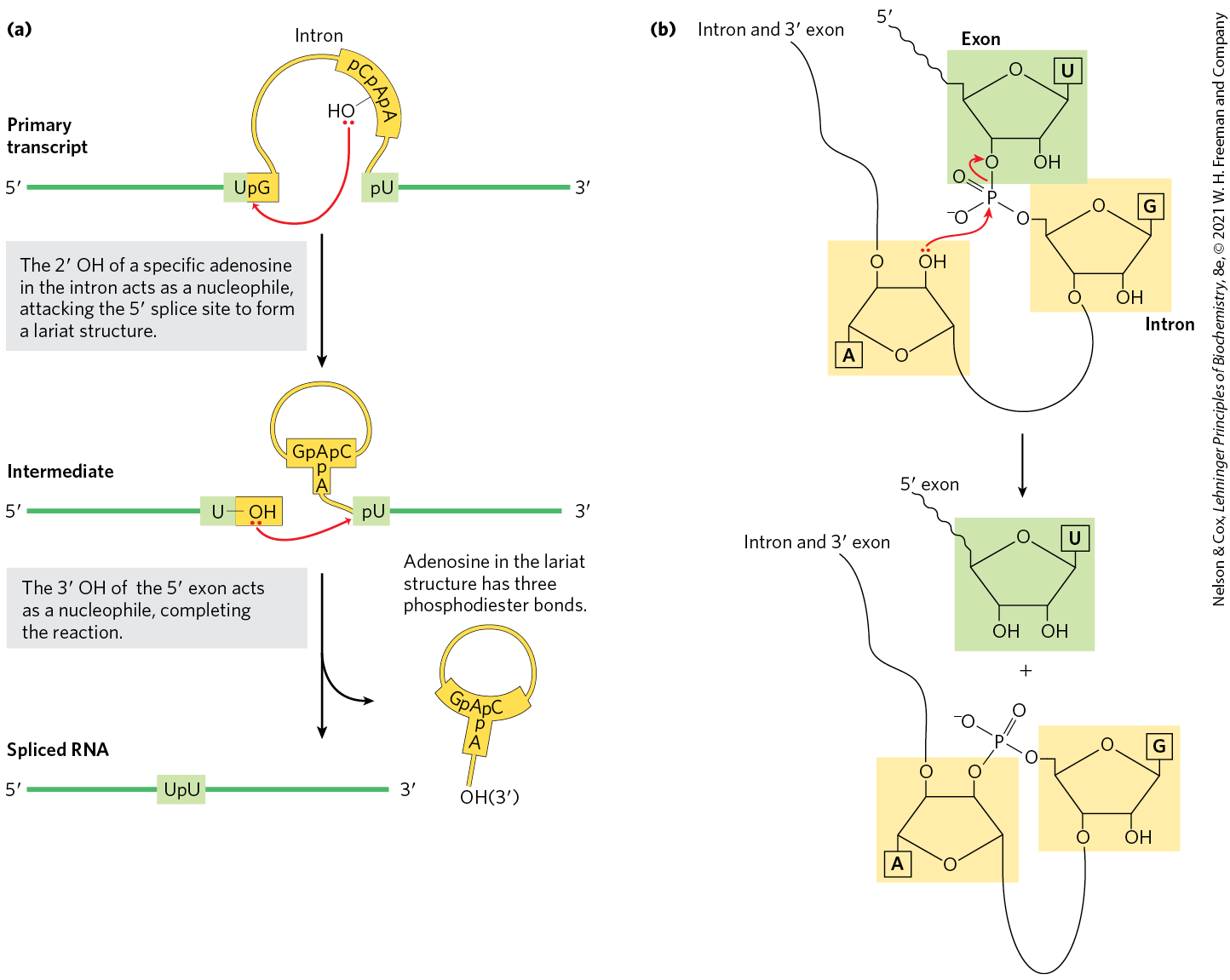
FIGURE 26-14 Splicing mechanism of group II introns. (a) In the first step, the OH of an internal A residue (called the branch point) attacks the phosphodiester bond at the splice site, resulting in splice site cleavage and lariat formation. In the second step, the free OH of the exon attacks the phosphodiester bond at the splice site, resulting in exon ligation and intron-lariat release. The spliceosome uses the same chemistry for intron removal, although different RNA sequences mark the intron boundaries and location of the branch point. (b) In the transesterification reaction that occurs during lariat formation, one phosphodiester bond is broken as a second one is created. This forms a lariatlike structure in which one branch is a ,-phosphodiester bond (the linkage between the intron branchpoint A and intron G nucleotides).
The group I splicing reaction requires a guanine nucleoside or nucleotide cofactor, but the cofactor is not used as a source of energy; instead, the -hydroxyl group of guanosine is used as a nucleophile in the first step of the splicing pathway. In group II splicing reactions, the nucleophile is the -hydroxyl group of an A residue within the intron (Fig. 26-14a). A branched lariat structure is formed as an intermediate (Fig. 26-14b). In both group I introns and group II introns, the hydroxyl of the exon that is displaced in the first step then acts as a nucleophile in a similar reaction at the end of the intron. The result is precise excision of the intron and ligation of the exons.
Thomas Cech
Self-splicing of introns was first revealed in 1982 in studies of the splicing mechanism of the group I rRNA intron from the ciliated protozoan Tetrahymena thermophila, conducted by Thomas Cech and colleagues. These workers transcribed isolated Tetrahymena DNA (including the intron) in vitro, using purified bacterial RNA polymerase. The resulting RNA spliced itself accurately without any protein enzymes from Tetrahymena. The discovery that RNAs could have catalytic functions was a milestone in our understanding of biological systems and a major step forward in the understanding of how life probably evolved. Catalytic RNAs like group I and group II introns share many features with protein-based enzymes, including folding into well-defined secondary and tertiary structures (Fig. 26-15). Catalytic RNAs and their significance in evolution are described in greater detail in Section 26.4.
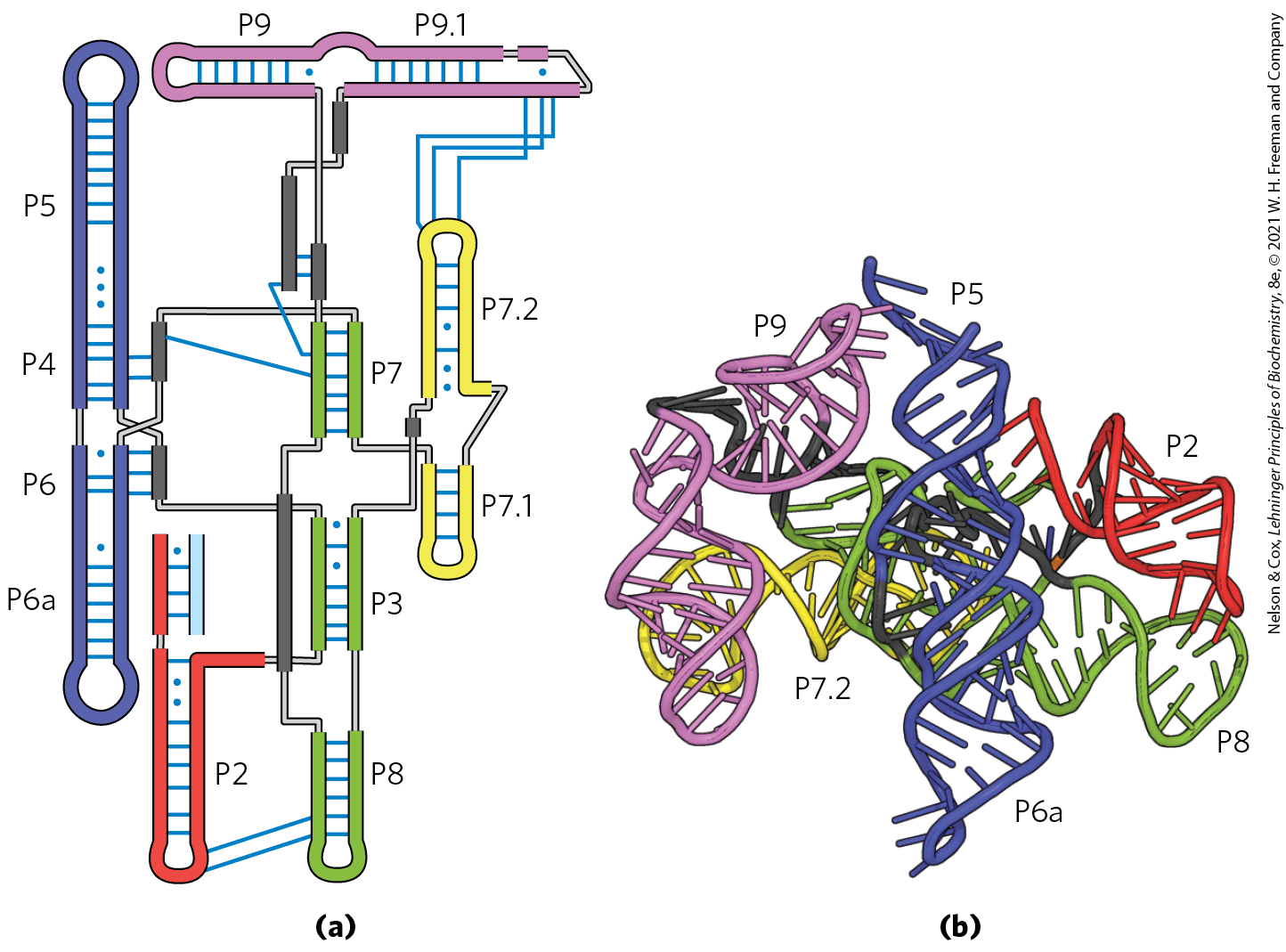
FIGURE 26-15 Structure of a group I intron. (a) The secondary structure of the group I intron ribozyme from phage Twort, a mycobacterium phage named for Frederick Twort, the physician who discovered phage in 1915. Like most catalytic RNAs, this intron adopts well-defined secondary structure. It is composed of multiple RNA duplexes (P2–P9, each differently colored) capped by hairpin structures. (b) The tertiary structure of the intron bound to a spliced RNA product, obtained by x-ray crystallography, shows that the RNA duplexes pack closely with one another to yield a compact ribozyme. The RNA duplexes are colored and named as in (a). [Data from PDB ID 1Y0Q, B. Golden et al., Nat. Struct. Biol. 12:82, 2005.]
In Eukaryotes the Spliceosome Carries out Nuclear pre-mRNA Splicing
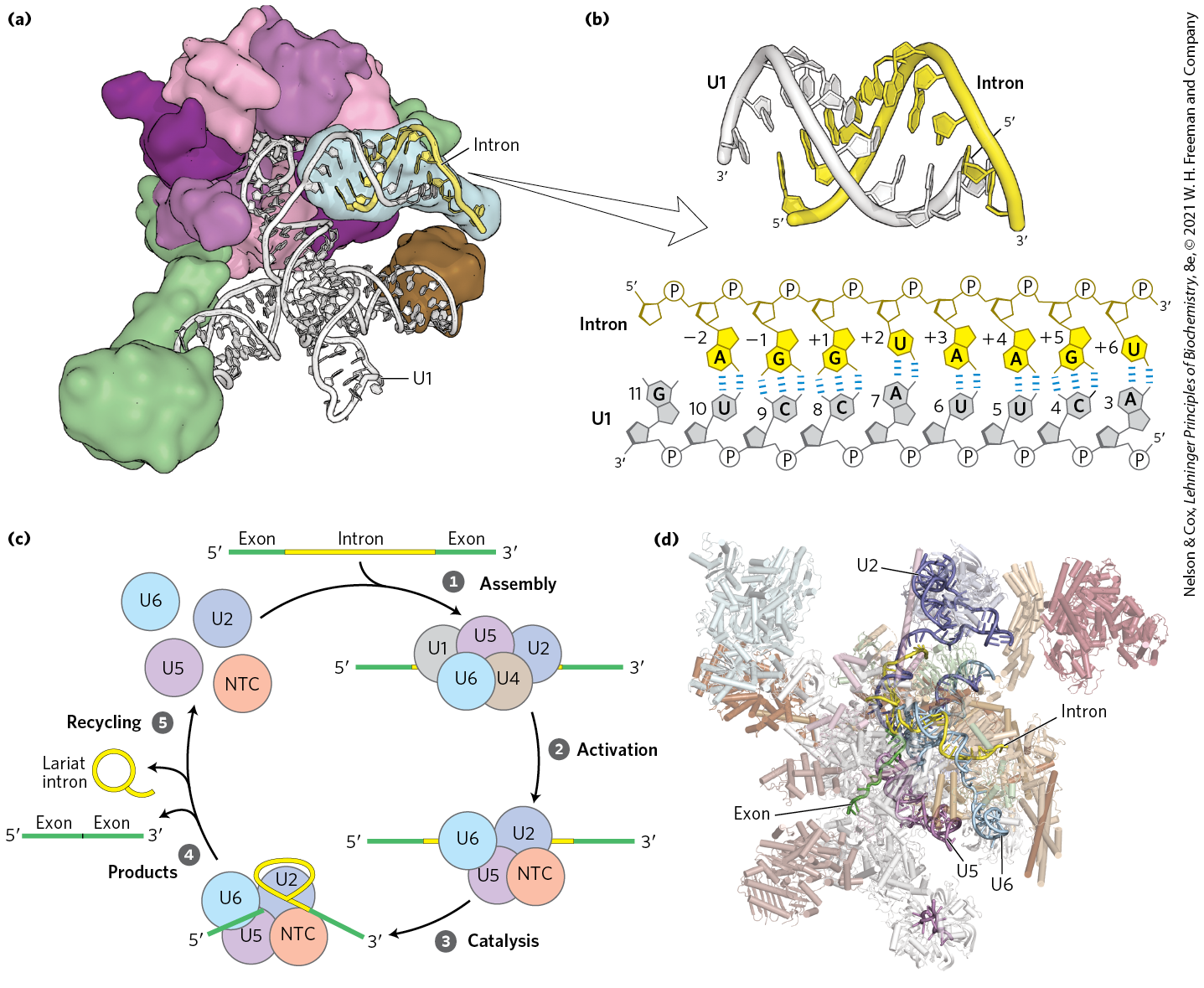
In eukaryotes, most introns undergo splicing by the same lariat-forming mechanism as the group II introns. However, the intron splicing takes place within a spliceosome, a large complex made up of multiple specialized RNP complexes called small nuclear ribonucleoproteins (snRNPs, pronounced snurps) and dozens of non-snRNP proteins. Each snRNP contains one of a class of eukaryotic RNAs, 100 to 200 nucleotides long, known as small nuclear RNAs (snRNAs). Five snRNAs (U1, U2, U4, U5, U6) involved in splicing reactions are generally found in abundance in eukaryotic nuclei. The U3 snRNP is also found in the nucleus but is involved in ribosome assembly and is not part of the spliceosome.
Joan Steitz
The role of snRNPs in the splicing reaction was discovered by Joan Steitz in a remarkable example of “bedside-to-bench” science. Using antibodies isolated from patients with autoimmune diseases, members of the Steitz laboratory were able to purify the spliceosome’s snRNP components and identify the associated snRNAs. Based on complementarity between the end of the U1 snRNA and the splice site of nuclear pre-mRNA introns (Fig. 26-16), Steitz proposed that snRNPs participate in the splicing reaction. Subsequently, it was discovered that patients suffering from the autoimmune disease lupus can generate antibodies against protein components of their own spliceosomes.
FIGURE 26-16 Processing of pre-mRNA primary transcripts by the spliceosome. (a) Small nuclear RNPs, such as the human U1 snRNP shown here, contain snRNAs associated with a number of proteins. The human U1 snRNP contains a single copy of the U1 snRNA and 10 associated polypeptides. (b) The U1 snRNP recognizes the splice site by base-pairing between the U1 snRNA and conserved RNA sequences within the intron that mark the exon/intron boundary. The sequences and structure shown were obtained from x-ray crystal structure shown in (a). The human U1 snRNA normally contains pseudouridines at positions 5 and 6; however, an unmodified RNA was used to determine this structure. (c) Spliceosomes are assembled on introns from snRNPs and proceed through stages of assembly, activation, catalysis of intron removal, release of the RNA products, and recycling of the splicing factors. In addition to the U snRNPs discussed in the text, a large protein-only supramolecular complex called the Prp19-containing complex (also known as NineTeen Complex, or NTC) is required for splicing and joins the spliceosome during the activation step. (d) Structure of the human spliceosome, determined by cryo-EM. Nuclear pre-mRNA splicing requires this large molecular machine composed of dozens of proteins and five snRNAs to remove many different introns. By comparison, using similar chemistry, group II introns can catalyze their own removal. In order to highlight the RNA catalytic core of the spliceosome and the U2, U5, and U6 snRNAs, some protein splicing factors are not shown in this view. The RNA appears discontinuous where the structure could not be resolved. [Data from (a, b) PDB ID 4PJO, Y. Kondo et al., eLife 4:e04986, 2015; (d) PDB ID 6QDV, S. Fica et al., Science 363:710, 2019.]
In yeast, the various snRNPs include about 80 different proteins, most of which have close homologs in all other eukaryotes. In humans, these conserved protein components are augmented by more than 200 additional proteins, which mostly participate in regulating the splicing reaction. Spliceosomes are thus among the most complex supramolecular machines in any eukaryotic cell. The RNA components of a spliceosome are the catalysts of the various splicing steps. The overall complex can be considered a highly flexible ribonucleoprotein enzyme that can adapt to the great diversity in size and sequence of nuclear pre-mRNAs.
Spliceosomal introns generally have the dinucleotide sequence GU at the end and AG at the end, marking the sites where splicing occurs. The U1 snRNA contains a sequence complementary to the splice site (Fig. 26-16b), and the U1 snRNP binds to this region by forming an RNA duplex with the pre-mRNA. A U2 snRNP binds to the end, also by base-pairing, and identifies the A residue that becomes the nucleophile used during the first transesterification reaction (Fig. 26-14). Addition of a complex of the U4, U5, and U6 snRNPs, called the tri-snRNP, leads to formation of the spliceosome (Fig. 26-16c).
Key parts of the splicing active site found in the U6 snRNA are initially sequestered by base-pairing to parts of U4 snRNA to prevent aberrant cleavage of nontarget phosphodiester bonds. In a process called activation, the U6 and U4 snRNAs must be unwound and separated to expose the active site needed for the first step in splicing. Unwinding of U4 and U6 as well as many other steps in splicing require ATP hydrolysis by a set of eight different ATPases that are part of the splicing machinery.
Spliceosomes are single turnover enzymes, meaning that each spliceosome can remove only one intron from a single transcript. As a result, spliceosomes undergo a complex cycle of assembly, activation, catalysis, product release, and recycling of the snRNP components each time an intron is removed (Fig. 26-16c, steps through ).
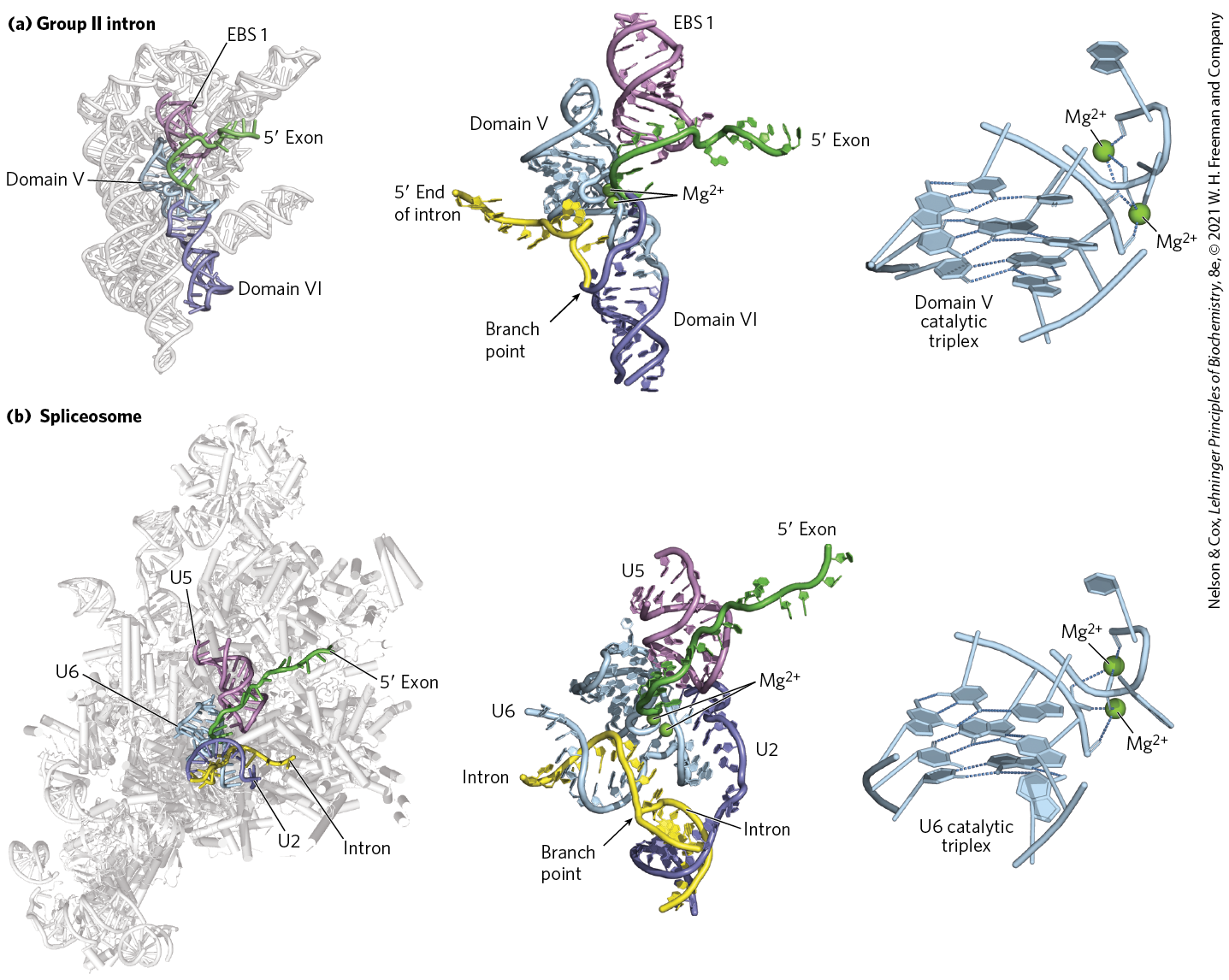
The chemical events of splicing — splice site cleavage by formation of an intron lariat followed by exon ligation — are identical in mechanism to that of group II introns, despite the former requiring dozens of proteins for activity and the latter being a self-splicing ribozyme. The similarities in chemistry as well as conservation between essential RNA components of each enzyme suggests that group II introns and spliceosomes are evolutionarily related to one another. Comparison of x-ray crystal structures of group II introns and cryo-EM structures of spliceosomes provide strong support for this hypothesis. Despite being surrounded by a large protein scaffold, the catalytic center of the spliceosome is composed of RNA and arranged in a nearly identical manner to that of group II introns (Fig. 26-17). Thus, the spliceosome uses a ribozyme core to carry out pre-mRNA splicing. As we shall see, some group II introns also contain domains that are themselves translated as mRNAs and encode proteins that bear striking similarity to those in the spliceosome, strengthening this evolutionary connection.
FIGURE 26-17 RNA active site conservation between group II introns and the spliceosome. A close-up examination of the active sites of (a) the Pylaiella littoralis group II intron and (b) the S. cerevisiae spliceosome reveal a similar arrangement of catalytic RNAs. In both cases, a catalytic RNA (domain V of the group II intron or U6 snRNA of the spliceosome) promotes a transesterification reaction by orienting the phosphodiester bond located at the splice site/intron junction for nucleophilic attack and by coordinating essential ions. In addition to similarities between domain V of group II introns and the U6 snRNA, group II intron domain VI and exon binding site 1 (EBS 1) play functional roles that are similar to those of the U2 and U5 snRNAs in the spliceosome. Close examination shows that the active sites of the group II intron and the spliceosome are nearly identical, formed by a complex arrangement of nucleotides called the catalytic triplex. The triplex is responsible for binding ions essential for catalysis as well as orienting the substrates for the splicing reaction. Conservation of sequence, chemistry, and three-dimensional structure suggests that the spliceosome evolved from a group II intron–like ribozyme. [Data from (a) left and center PDB ID 4R0D, A. R. Robart et al., Nature 514:193, 2014; right PDB ID 6QDV, S. M. Fica et al., Science 363:710, 2019; (b) left PDB ID 5LJ3, W. P. Galej et al., Nature 537:197, 2016; center PDB ID 5MQ0, S. M. Fica et al., Nature 542:377, 2017; right PDB ID 6ME0, D. B. Haack et al., Cell 178:612, 2019. Information from W. Galej et al., Chem. Rev. 118:4156, 2018.]
About 1% of human introns are spliced by a less common type of spliceosome, called the minor spliceosome, in which the U1, U2, U4, and U6 snRNPs are replaced by the U11, U12, U4atac, and U6atac snRNPs. Whereas U1- and U2-containing spliceosomes remove introns with ()GU and AG() terminal sequences, the minor spliceosomes remove a rare class of introns that have ()AU and AC() terminal sequences to mark the splice sites. Introns removed by either the major or the minor spliceosome most often remain in the nucleus and are degraded.
Some components of the splicing apparatus are tethered to the CTD of RNA polymerase II, indicating that splicing, like other RNA processing reactions, is tightly coordinated with transcription. Most splicing in humans occurs cotranscriptionally, meaning that splicing occurs while Pol II is still transcribing the gene. For this to occur correctly, the rates of transcription, capping, splicing, and end formation must be carefully regulated. Splicing of a pre-mRNA in the nucleus can also have profound effects on the function of the mRNA in the cytoplasm. Lynne Maquat and Melissa Moore discovered that the human spliceosome leaves behind a set of proteins on each spliced mRNA near the junction between two exons. This exon junction complex is retained on the mRNA as it is exported to the cytoplasm, where it can regulate the extent to which an mRNA can be translated over its lifetime before degradation.
Proteins Catalyze Splicing of tRNAs
A fourth and final class of introns, found in certain tRNAs and a few mRNAs such as XBP1, is distinguished from other intron types in that the splicing reaction requires endonucleases and ligases made of protein and does not involve catalytic RNAs. The splicing endonuclease cleaves the phosphodiester bonds at both ends of the intron, and the two exons are joined by a mechanism similar to the DNA ligase reaction (see Fig. 25-15).
Eukaryotic mRNAs Have a Distinctive End Structure
At their end, most eukaryotic mRNAs undergoing translation in the cell cytoplasm have a string of A residues, about 30 residues in yeast and 50 to 100 in animals, called the poly(A) tail. This tail serves as a binding site for one or more specific proteins. The poly(A) tail and its associated proteins have a variety of roles in coordinating transcription and translation, and may help protect mRNA from enzymatic destruction. Many bacterial mRNAs also acquire poly(A) tails, but these tails stimulate decay of mRNA rather than protecting it from degradation.
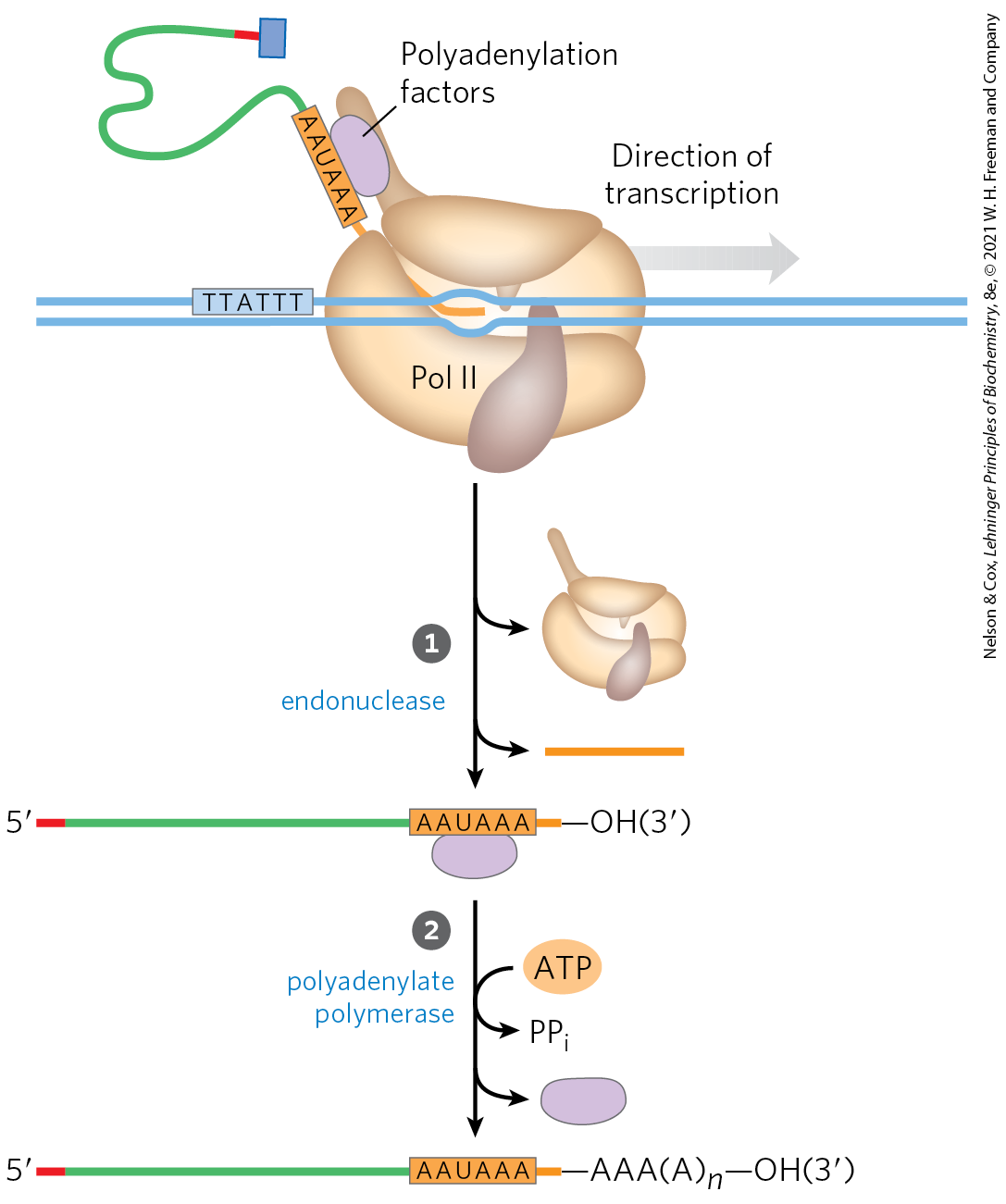
The poly(A) tail is added in a multistep process. The transcript is extended beyond the site where the poly(A) tail is to be added, then is cleaved at the poly(A) addition site by an endonuclease component of a large enzyme complex, again associated with the CTD of RNA polymerase II (Fig. 26-18). The mRNA site where cleavage occurs is marked by two sequence elements: the highly conserved sequence ()AAUAAA(), 10 to 30 nucleotides on the side (upstream) of the cleavage site, and a less well-defined sequence rich in G and U residues, 20 to 40 nucleotides downstream of the cleavage site. Cleavage generates the free -hydroxyl group that defines the end of the mRNA, to which A residues are immediately added by polyadenylate polymerase, which catalyzes the reaction
FIGURE 26-18 Addition of the poly(A) tail to the primary RNA transcript of eukaryotes. Pol II synthesizes RNA beyond the segment of the transcript containing the cleavage signal sequences, including the highly conserved upstream sequence ()AAUAAA. This cleavage signal sequence is bound by an enzyme complex that includes an endonuclease, a polyadenylate polymerase, and several other multisubunit proteins involved in sequence recognition, stimulation of cleavage, and regulation of the length of the poly(A) tail; all of these proteins are tethered to the CTD. The RNA is cleaved by the endonuclease at a point 10 to 30 nucleotides to (downstream of) the sequence AAUAAA. The polyadenylate polymerase synthesizes a poly(A) tail 80 to 250 nucleotides long, beginning at the cleavage site.
where to 250. This enzyme does not require a template but does require the cleaved mRNA as a primer. These longer poly(A) tails are added in the nucleus, and then shortened significantly after the mRNA is transported to the cytoplasm.
The overall processing of a typical eukaryotic mRNA is summarized in Figure 26-19. In some cases, the polypeptide-coding region of the mRNA is also modified by RNA “editing” (see Section 27.1 for details). This editing includes processes that add or delete bases in the coding regions of primary transcripts or that change the sequence (such as by enzymatic deamination of a C residue to create a U residue). A particularly dramatic example occurs in trypanosomes, which are parasitic protists: large regions of an mRNA are synthesized without any uridylate, and the U residues are inserted later by RNA editing.
FIGURE 26-19 Overview of the processing of a eukaryotic mRNA. The ovalbumin gene, shown here, has introns A to G and exons 1 to 7 and L (L encodes a signal peptide sequence that targets the protein for export from the cell; see Fig. 27-38). About three-quarters of the RNA is removed during processing. Pol II extends the primary transcript well beyond the cleavage and polyadenylation site (“extra RNA”) before terminating transcription.
A Gene Can Give Rise to Multiple Products by Differential RNA Processing
One of the paradoxes of modern genomics is that the apparent complexity of organisms does not correlate with the number of protein-coding genes, or even the amount of genomic DNA. Some eukaryotic mRNA transcripts can be processed in more than one way to produce different mRNAs and thus different polypeptides. Much of the variability in processing is the result of alternative splicing, in which a particular exon may or may not be incorporated into the mature mRNA transcript. Alternative splicing occurs in a relatively small number of transcripts in yeast, but in more than 95% of human genes. Changes in alternative splicing can have profound impact on the development of an organism (Box 26-2). Alternative splicing of a single transcription factor in the staple grain quinoa differentiates palatable sweet varieties from those too bitter to ingest without processing. In Drosophila, sex is determined by alternative splicing of the sex lethal (Sxl) transcript based on the number of X chromosomes present in the cell.
Figure 26-20a illustrates how alternative splicing patterns can produce more than one protein from a common pre-mRNA. The pre-mRNA contains molecular signals for all the alternative processing pathways, and the pathway favored in a given cell or metabolic situation is determined by processing factors, RNA-binding proteins that promote one particular path. For example, splicing regulatory proteins or heterogeneous ribonuclear proteins (hnRNPs) may bind these signals and promote or inhibit spliceosome assembly at that site. There are many additional patterns of alternative splicing.
FIGURE 26-20 Alternative transcript production in eukaryotes. (a) Alternative splicing patterns. Two splice sites are shown. Different mature mRNAs are produced from the same primary transcript. (b) Two alternative cleavage and polyadenylation sites, and .
Complex transcripts can also have more than one site where poly(A) tails can form (Fig. 26-20b). If there are two or more sites for cleavage and polyadenylation, use of the one closest to the end will remove more of the primary transcript sequence. This mechanism, called poly(A) site choice, generates diversity in the variable domains of immunoglobulin heavy chains (see Fig. 25-42).
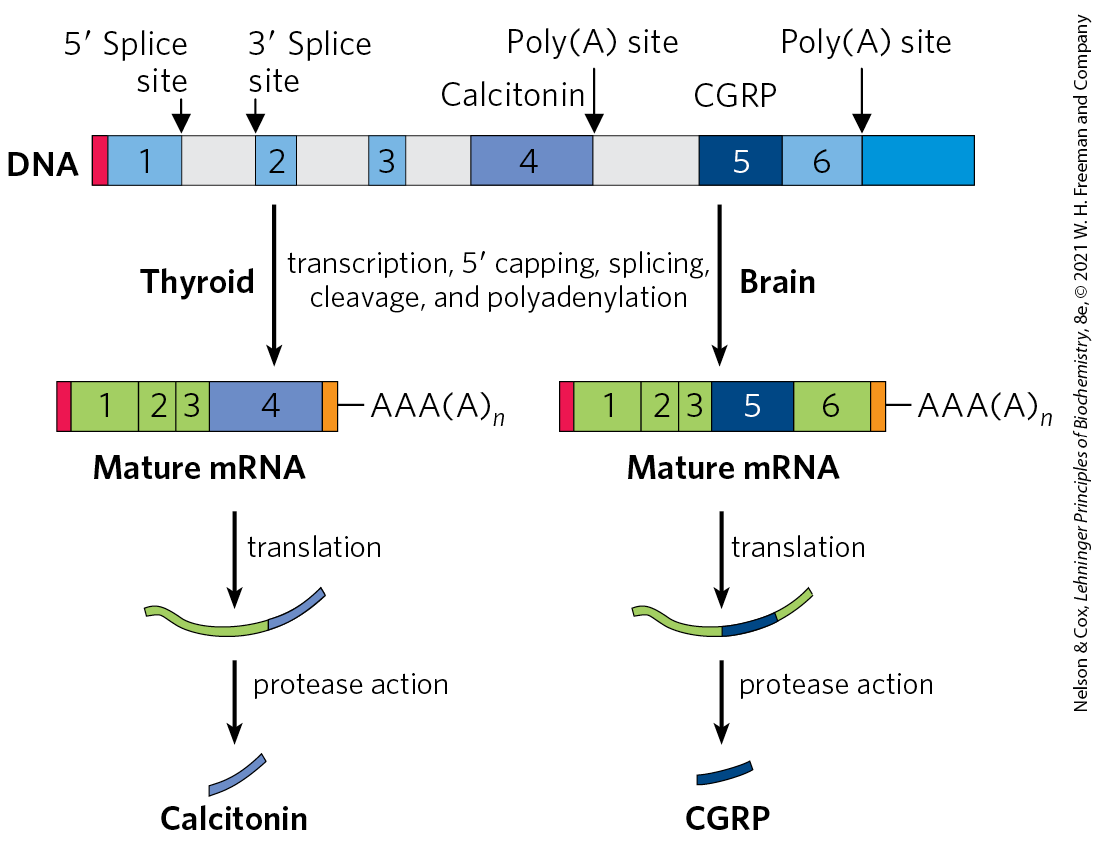
Both alternative splicing and poly(A) site choice come into play in the expression of many genes. For example, a single RNA transcript is processed using both mechanisms to produce two different hormones: the calcium-regulating hormone calcitonin in rat thyroid and calcitonin-gene-related peptide (CGRP) in rat brain (Fig. 26-21). Together, alternative splicing and poly(A) site choice greatly increase the variety of proteins generated from the genomes of higher eukaryotes.
FIGURE 26-21 Alternative processing of the calcitonin gene transcript in rats. The calcitonin gene encodes a primary transcript with two poly(A) sites; one predominates in the brain, the other in the thyroid. In the brain, splicing eliminates the calcitonin exon (exon 4); in the thyroid, this exon is retained. The resulting peptides are processed further to yield the final hormone products: calcitonin in the thyroid and calcitonin-gene-related peptide (CGRP) in the brain.
Ribosomal RNAs and tRNAs Also Undergo Processing
Posttranscriptional processing is not limited to mRNA. Ribosomal RNAs of bacterial, archaeal, and eukaryotic cells are made from longer precursors called pre-ribosomal RNAs, or pre-rRNAs. Transfer RNAs are similarly derived from longer precursors. These RNAs may also contain a variety of modified nucleosides; some examples are shown in Figure 26-22.
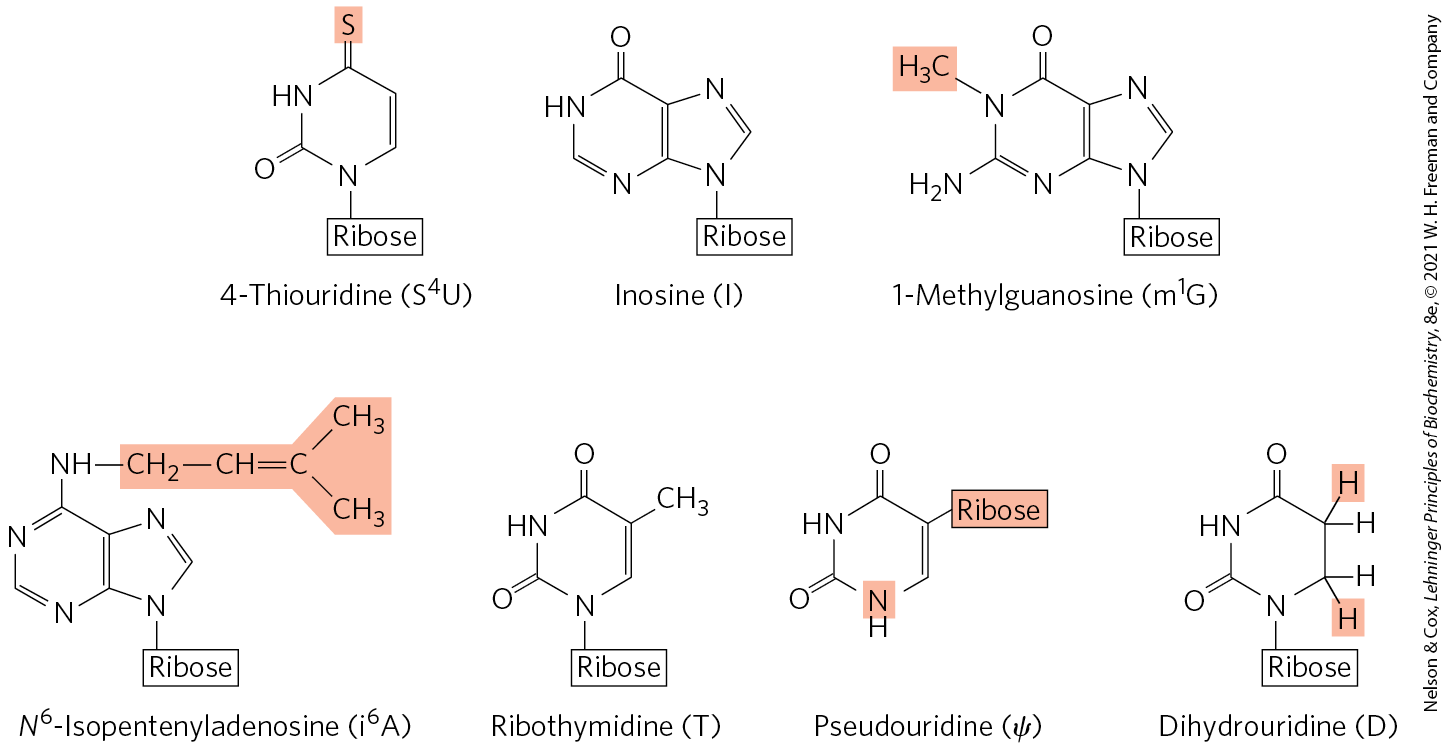
FIGURE 26-22 Some modified bases of RNA produced in posttranscriptional reactions. The standard symbols are shown in parentheses. This is just a small sampling of the 96 modified nucleosides known to occur in different RNA species, with 81 different types known in tRNAs and 30 observed to date in rRNAs. Notice the unusual ribose attachment point in pseudouridine. A complete listing of these modified bases can be found in the Modomics database of RNA modification pathways (http://iimcb.genesilico.pl/modomics/).
Ribosomal RNAs
In bacteria, 16S, 23S, and 5S rRNAs (and some tRNAs, although most tRNAs are encoded elsewhere) arise from a single 30S RNA precursor of about 6,500 nucleotides. RNA at both ends of the 30S precursor and segments between the rRNAs are removed during processing (Fig. 26-23). The 16S and 23S rRNAs contain modified nucleosides. In E. coli, the 11 modifications in the 16S rRNA include a pseudouridine and 10 nucleosides methylated on the base or the -hydroxyl group or both. The 23S rRNA has 10 pseudouridines, 1 dihydrouridine, and 12 methylated nucleosides. In bacteria, each modification is generally catalyzed by a distinct enzyme. Methylation reactions use S-adenosylmethionine as cofactor. No cofactor is required for pseudouridine formation.
FIGURE 26-23 Processing of pre-rRNA transcripts in bacteria. Before cleavage, the 30S RNA precursor is methylated at specific bases (red tick marks), and some uridine residues are converted to pseudouridine (blue tick) or dihydrouridine (black tick) residues. The methylation reactions are of multiple types, some occurring on bases and some on -hydroxyl groups. Cleavage liberates precursors of rRNAs and tRNA(s). Cleavage at the points labeled 1, 2, and 3 is carried out by the enzymes RNase III, RNase P, and RNase E, respectively. As discussed later in the text, RNase P is a ribozyme. The final 16S, 23S, and 5S rRNA products result from the action of a variety of specific nucleases. The seven copies of pre-rRNA gene in the E. coli chromosome differ in the number, location, and identity of tRNAs included in the primary transcript. Some copies of the gene have additional tRNA gene segments between the 16S and 23S rRNA segments and at the far end of the primary transcript.
The genome of E. coli encodes seven pre-rRNA molecules. All of these genes have essentially identical rRNA-coding regions, but they differ in the segments between these regions. The segment between the 16S and 23S rRNA genes generally encodes one or two tRNAs, with different tRNAs produced from different pre-rRNA transcripts. Coding sequences for tRNAs are also found on the side of the 5S rRNA in some precursor transcripts.
The situation in eukaryotes is even more complicated (see Fig. 27-17). The entire process is initiated in the nucleolus, in large complexes that assemble on the rRNA precursor as it is synthesized by Pol I. There is a tight coupling between rRNA transcription, rRNA maturation, and ribosome assembly in the nucleolus. Each complex includes the ribonucleases that cleave the rRNA precursor, the enzymes that modify particular bases, large numbers of ncRNAs called small nucleolar RNAs, or snoRNAs, that guide nucleoside modification and some cleavage reactions, and ribosomal proteins. In yeast, the entire process involves the pre-rRNA, more than 170 nonribosomal proteins, snoRNAs for each nucleoside modification (about 70, because some snoRNAs guide two types of modification), and the 78 ribosomal proteins. Humans have an even greater number of modified nucleosides, about 200, and a greater number of associated snoRNAs. The composition of the complexes may change as the ribosomes are assembled, and many of the intermediate complexes rival the ribosome itself in complexity. The 5S rRNA of most eukaryotes is made as a completely separate transcript by a different polymerase (Pol III).
The most common nucleoside modifications in eukaryotic rRNAs are conversion of uridine to pseudouridine and adoMet-dependent nucleoside methylation (often at -hydroxyl groups). These reactions often rely on snoRNA-protein complexes, or snoRNPs, each consisting of a snoRNA and four or five proteins, including the enzyme that carries out the modification. There are two classes of snoRNPs, both defined by key conserved sequence elements referred to as lettered boxes. The box H/ACA snoRNPs function in pseudouridylylation, and box C/D snoRNPs in -O-methylations. The snoRNAs are 60 to 300 nucleotides long. Each snoRNA includes a 10 to 21 nucleotide sequence that is perfectly complementary to some site on an rRNA and serves to identify the modification site (Fig. 26-24). The conserved sequence elements in the remainder of the snoRNA fold into structures that are bound by the snoRNP proteins.
FIGURE 26-24 RNA pairing with box H/ACA snoRNAs to guide pseudouridylations. The pseudouridine conversion sites in the target rRNA are in the regions paired with the snoRNA, and the conserved H/ACA box sequences are protein-binding sites. [Information from T. Kiss, Cell 109:145, 2002.]
Transfer RNAs
Most cells have 40 to 50 distinct tRNAs, and eukaryotic cells have multiple copies of many of the tRNA genes. Transfer RNAs are derived from longer RNA precursors by enzymatic removal of nucleotides from the and ends (Fig. 26-25). In eukaryotes, introns are present in a few tRNA transcripts and must be excised. Where two or more different tRNAs are contained in a single primary transcript, they are separated by enzymatic cleavage. The endonuclease RNase P, found in all organisms, removes RNA at the end of tRNAs. This enzyme contains both protein and RNA. The RNA component is essential for activity, and in bacterial cells it can carry out its processing function with precision even without the protein component. RNase P is another example of a catalytic RNA, as described in more detail below. The end of tRNAs is processed by one or more nucleases, including the exonuclease RNase D.
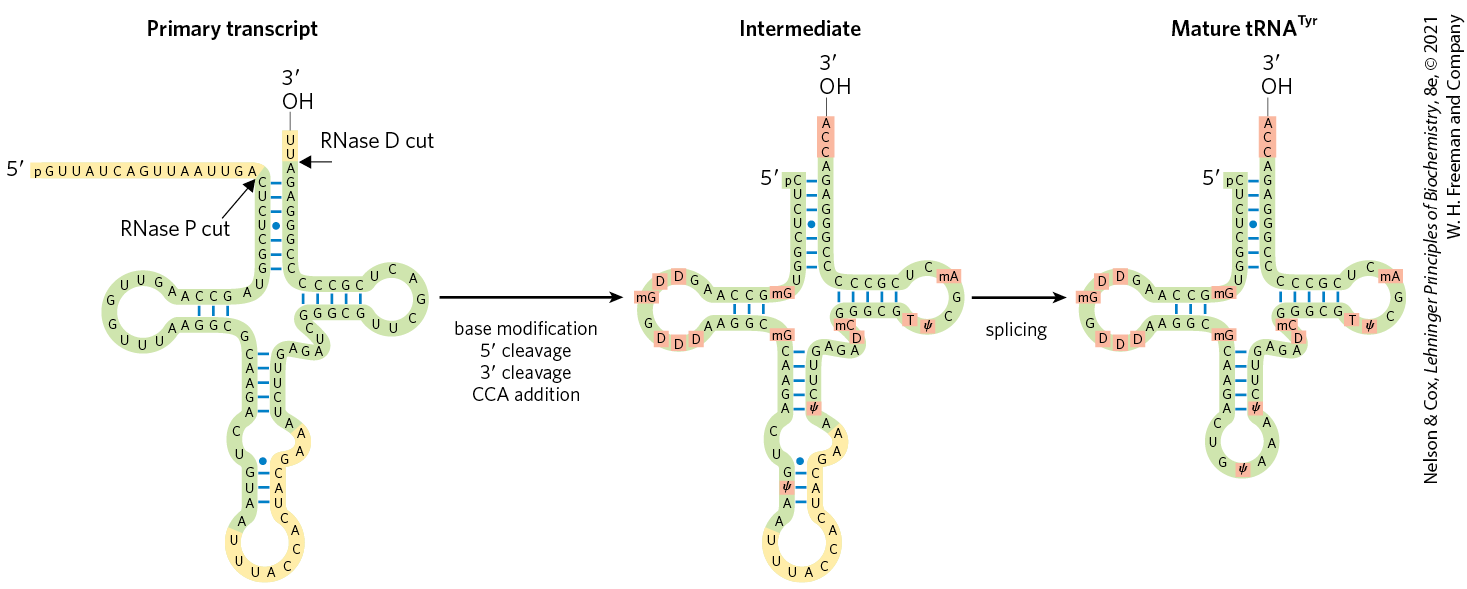
FIGURE 26-25 Processing of tRNAs in bacteria and eukaryotes. The yeast tRNATyr (the tRNA specific for tyrosine binding; see Chapter 27) is used to illustrate the important steps. Short blue lines represent normal base pairing; blue dots indicate G–U base pairs. The nucleotide sequences shown in yellow are removed from the primary transcript. The ends are processed first, the end before the end. CCA is then added to the end, a necessary step in processing all eukaryotic tRNAs and for those bacterial tRNAs that lack this sequence in the primary transcript. While the ends are being processed, specific bases in the rest of the transcript are modified (see Fig. 26-22). For the eukaryotic tRNA shown here, the final step is splicing of the 14 nucleotide intron by a protein enzyme. Introns are found in some eukaryotic tRNAs but not in bacterial tRNAs.
Transfer RNA precursors may undergo further posttranscriptional processing. The -terminal trinucleotide CCA() to which an amino acid is attached during protein synthesis (Chapter 27) is absent from some bacterial tRNA precursors and all eukaryotic tRNA precursors and is added during processing (Fig. 26-25). This addition is carried out by tRNA nucleotidyltransferase, an unusual enzyme that binds the three ribonucleoside triphosphate precursors in separate active sites and catalyzes formation of the phosphodiester bonds to produce the CCA() sequence. The creation of this defined sequence of nucleotides is therefore not dependent on a DNA or RNA template — the template is the binding site of the enzyme.
The final type of tRNA processing is the modification of some bases by methylation, deamination, or reduction (Fig. 26-22). These modifications can change how the tRNA interacts with cellular proteins and even how the tRNA is used by the ribosome during translation. In the case of pseudouridine, the base (uracil) is removed and reattached to the sugar through C-5. Some of these modified bases occur at characteristic positions in all tRNAs (Fig. 26-25).
Special-Function RNAs Undergo Several Types of Processing
The number of known classes of special-function noncoding RNAs (ncRNAs) is expanding rapidly, as is the variety of functions known to be associated with them. Many of these ncRNAs also undergo processing.
The snRNAs and snoRNAs not only facilitate RNA processing reactions but also are themselves synthesized as larger precursors and then processed. Many snoRNAs are encoded within the introns of other genes. As the introns are spliced from the pre-mRNA, proteins bind to the snoRNA sequences and ribonucleases remove the extra RNA at the and ends to form the snoRNP. The snRNAs destined for spliceosomes are synthesized as pre-snRNAs, and ribonucleases remove the extra RNA at each end. Particular nucleosides in snRNAs are also subject to 11 types of modification, with -O-methylation and conversion of uridine to pseudouridine predominating.
MicroRNAs (miRNAs) are a special class of noncoding RNAs involved in gene regulation. The miRNAs are about 22 nucleotides long, complementary in sequence to particular regions of mRNAs. Found in plants and in animals, from worms to mammals, they promote mRNA degradation and suppress translation to fine-tune gene expression. About 1,500 human genes encode miRNAs, and one or more of these miRNAs affect the expression of most protein-coding genes.
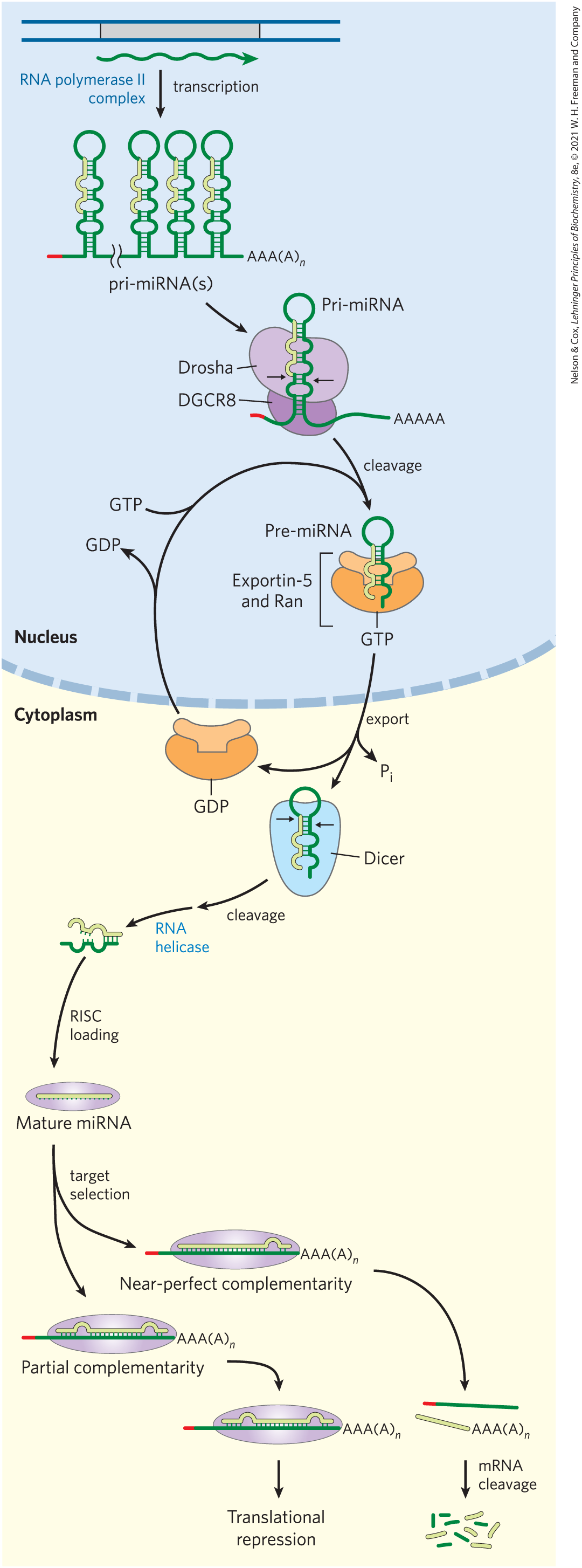
The miRNAs are synthesized from much larger precursors, in several steps (Fig. 26-26). The primary transcripts for miRNAs (pri-miRNAs) vary greatly in size; some are encoded in the introns of other genes and are coexpressed with these host genes. Processing of pri-miRNA is mediated by two endoribonucleases in the RNase III family, Drosha and Dicer. First, in the nucleus, the pri-miRNA is reduced to a 70 to 80 nucleotide precursor miRNA (pre-miRNA) by a protein complex including Drosha and another protein, DGCR8. The pre-miRNA is then exported to the cytoplasm in a complex with two proteins, exportin-5 and the Ran GTPase (see Fig. 27-42). In the cytoplasm, Ran hydrolyzes the GTP, then exportin-5 and the pre-miRNA are released. The pre-miRNA is then acted on by Dicer to produce the nearly mature miRNA paired with a short RNA complement. The complement is removed by an RNA helicase, and the mature miRNA is incorporated into protein complexes, such as the RNA-induced silencing complex (RISC), which then bind a target mRNA. If the complementarity between miRNA and its target is nearly perfect, the target mRNA is cleaved. If the complementarity is only partial, the complex blocks translation of the target mRNA. The roles of miRNAs and RISC in gene regulation are detailed in Chapter 28.
FIGURE 26-26 Synthesis and processing of miRNAs. The primary transcript of miRNAs is a larger RNA of variable length, called pri-miRNA. The pri-miRNA undergoes a number of processing events both in the nucleus and in the cytoplasm to make a mature miRNA. Once the miRNA has been loaded into a protein complex called RISC, it can then hybridize to mRNAs and repress their translation or trigger their cleavage and destruction. [Information from E. Wienholds and R. H. A. Plasterk, FEBS Lett. 579:5911, 2005; V. N. Kim et al., Nat. Rev. Mol. Cell Biol. 10:126, 2009, Figs 2–4.]
Cellular mRNAs Are Degraded at Different Rates
The expression of genes is regulated at many levels. A crucial factor governing a gene’s expression is the cellular concentration of its associated mRNA. The concentration of any molecule depends on two factors: its rate of synthesis and its rate of degradation. When synthesis and degradation of an mRNA are balanced, the concentration of the mRNA remains in a steady state. A change in either rate will lead to net accumulation or depletion of the mRNA. Degradative pathways ensure that mRNAs do not build up in the cell and direct the synthesis of unnecessary proteins.
The rates of degradation vary greatly for mRNAs from different eukaryotic genes. For a gene product that is needed only briefly, the half-life of its mRNA may be only minutes or even seconds. Gene products needed constantly by the cell may have mRNAs that are stable over many cell generations. The average half-life of the mRNAs of a vertebrate cell is about 3 hours, with the pool of each type of mRNA turning over about 10 times per cell generation. The half-life of bacterial mRNAs is much shorter — only about 1.5 min — perhaps because of regulatory requirements.
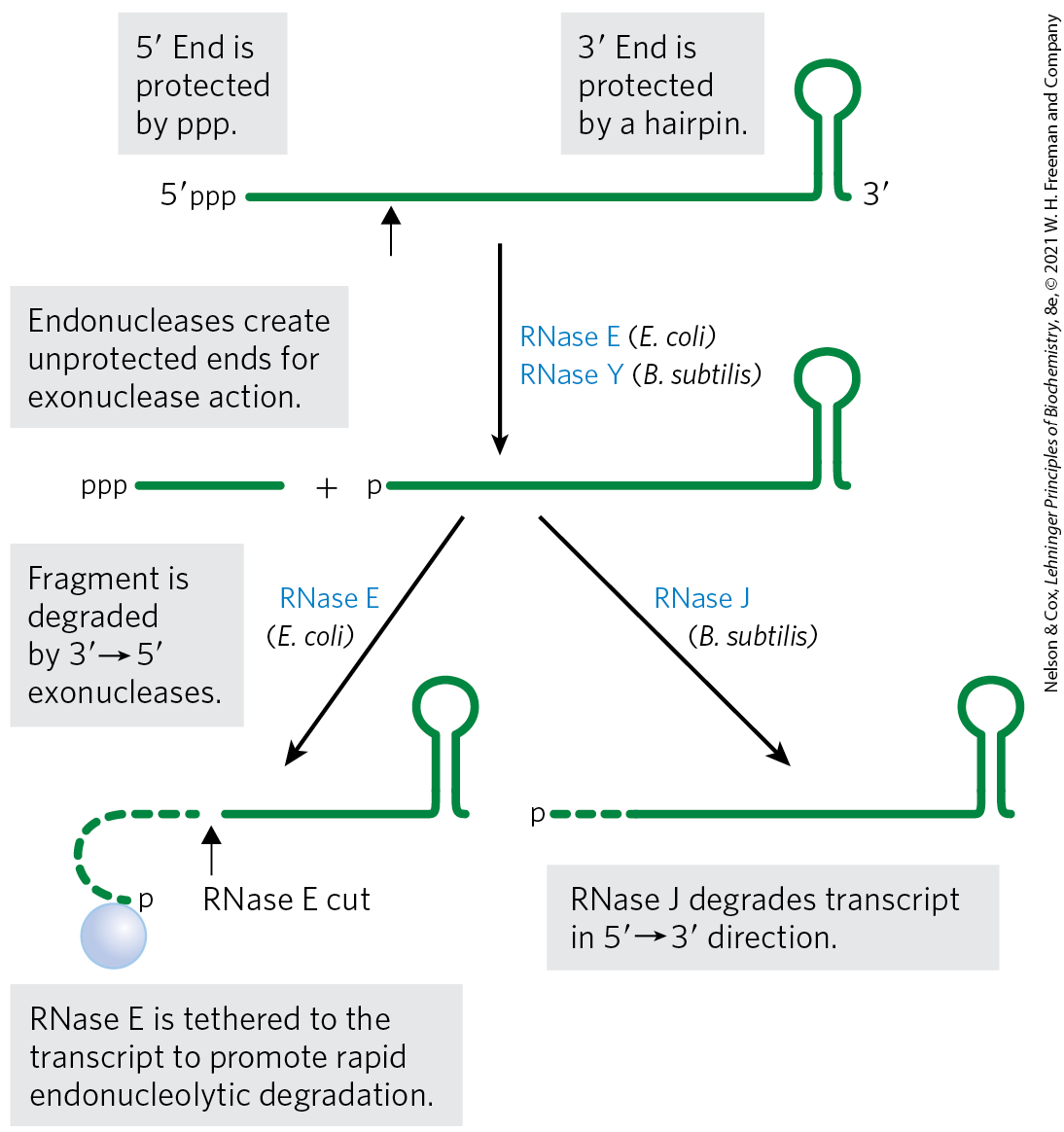
Messenger RNA is degraded by ribonucleases present in all cells. In E. coli, mRNAs typically contain triphosphates remaining from the initiation of transcription. These groups protect the mRNA from degradation. As a result, mRNA decay begins with one or several cuts by an endoribonuclease, followed by degradation by exoribonucleases (Fig. 26-27). The initial cut by the endonuclease generates an RNA fragment with a monophosphate end, which serves to tether the endonuclease to the transcript and ensure its rapid destruction. Some bacteria (Bacillus subtilis, for example), have exonucleases that also recognize the monophosphate end and can degrade RNA fragments in the direction.
FIGURE 26-27 Degradation of RNA in bacteria. In bacteria, mRNA degradation usually begins by endonucleolytic cleavage because the and ends of the mRNA are often protected by a triphosphate and hairpin structure, respectively. In E. coli, the RNase E endonuclease carries out this cleavage, whereas in B. subtilis the cleavage is carried out by the RNase Y endonuclease. The endonuclease activity produces RNA fragments that serve as substrates for or exonucleases. All bacteria contain exonucleases such as PNPase, RNase R, or RNase II. Some species, like B. subtilis, also contain a exonuclease called RNase J. The phosphate produced by the endonuclease after the first cleavage can also serve as a tether to link the RNase E endonuclease directly to the mRNA, ensuring its rapid destruction. [Information from M. Hui et al. Annu. Rev. Genet. 48:537, 2014.]
Polynucleotide phosphorylase (PNPase) is a common exoribonuclease responsible for the degradation of many mRNAs in bacteria, chloroplasts, and mitochondria. It catalyzes the reversible phosphorolysis (rather than hydrolysis) of the mRNA chain using orthophosphate as the nucleophile. The PNPase reaction is readily reversible and the enzyme can also add nucleotides to the ends of bacterial mRNAs. Decay of mRNAs containing complex end structures, such as the hairpins responsible for -independent transcription termination (see Fig. 26-7), can involve multiple rounds of lengthening and shortening of the mRNA by PNPase until it is finally consumed. This unusual nontemplated RNA polymerization activity of PNPase proved to be critical for production of mRNA polymers used for deciphering the genetic code (Chapter 27).
As we have previously seen with transcription and RNA processing, the analogous processes for RNA degradation in eukaryotes are much more complex than their bacterial counterparts. Eukaryotes have multiple pathways for mRNA decay, and the pathway used can depend on the mRNA location, its structure, its association with ribosomes, and other factors. However, in most cases, decapping the end and shortening the poly(A) tail are critical steps for allowing exonucleases to access the mRNA.
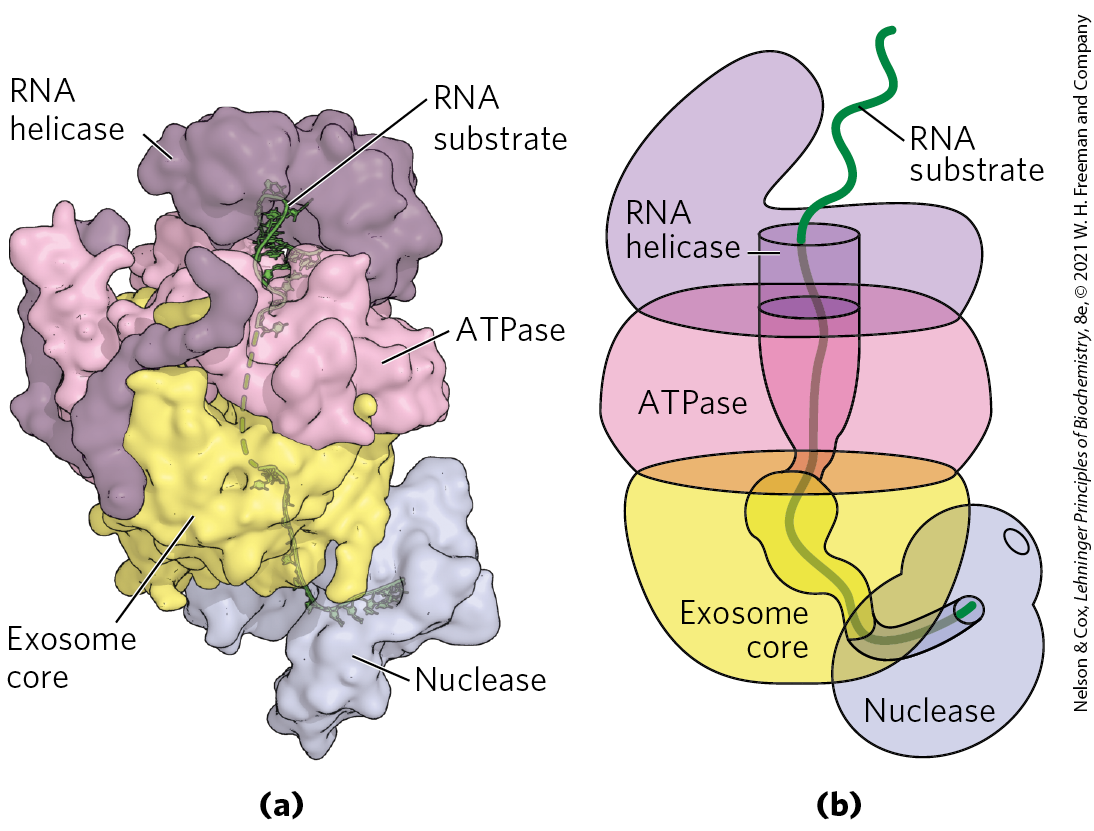
All eukaryotes also have large exoribonucleases called exosomes, which are responsible for the degradation for nearly all types of RNA. Exosomes are multisubunit complexes containing about 10 proteins. Specialized exosomes exist in the nucleus, cytoplasm, and nucleolus. The core of the exosome is a barrel-like structure through which RNA is threaded (Fig. 26-28). Even though this core is structurally similar to bacterial PNPase, RNA is not degraded within the barrel. Instead, the barrel serves as an adapter that efficiently channels the RNA to associated enzymes with exonuclease and endonuclease activity.
FIGURE 26-28 Essential role of the exosome in eukaryotic RNA degradation. (a) Exosomes are multisubunit enzymes in which RNA is threaded through a central barrel and fed into a nuclease. In this structure, the exosome core is topped by ATPase and RNA helicase modules that help unwind RNA secondary structures so that single-stranded RNA can pass into the core. Below the core is a nuclease responsible for cleaving the RNA. (b) In this cartoon schematic, the passage of the substrate RNA through the barrel-like exosome and to the nuclease is highlighted. [Data from PDB ID 4IFD, D. L. Makino et al., Nature 495:70, 2013; PDB ID 4OO1, E. V. Wasmuth et al., Nature 511:435, 2014. Information from K. Januszyk and C. D. Lima, Curr. Opin. Struct. Biol. 24:132, 2014.]
SUMMARY 26.2 RNA Processing
Many primary transcripts produced in bacteria and eukaryotes must be processed into a mature form to be functional. Processing can include modifications to the and ends of the RNA, removal of internal RNA sequences by splicing, and modifications of the RNA nucleotides.
Eukaryotic mRNAs have an inverted 7-methylguanosine residue cap at their end. The cap helps to protect the RNA from degradation and interacts with proteins important for cellular transport and translation.
Many organisms contain genes in which the coding information is interrupted by introns. Splicing removes these introns and joins the flanking exons. Nearly every human gene contains multiple introns, which can vary dramatically in size.
There are four classes of introns: group I, group II, spliceosome-processed introns, and protein-processed introns. Group I and II introns are self-splicing with RNAs capable of carrying out catalysis independent of protein enzymes. Catalytic RNAs share features in common with protein-based enzymes.
Nuclear-encoded introns in eukaryotes are removed by a large RNP machine called a spliceosome. A spliceosome is a single-turnover enzyme containing snRNA and protein that recognizes introns by base-pairing with the snRNAs. Even though a spliceosome contains dozens of proteins, it uses an RNA active site and mechanism similar to that of group II introns.
Some tRNAs and a few mRNAs contain introns that must be removed by protein-based endonuclease and ligase enzymes.
In eukaryotes, transcription terminates when an endonuclease cleaves the nascent RNA, freeing it from Pol II. A poly(A) tail is then added to the end of the RNA by a polyadenylate polymerase.
Alternative splicing and alternative poly(A) site choice in eukaryotes allow for many different transcripts to be produced from a single gene.
The primary transcripts of tRNAs, rRNAs, and miRNAs also undergo extensive processing, including endonucleolytic cleavage and chemical modification. Correct placement of these modifications is often guided by snoRNAs that base-pair with the target RNA.
The cellular lifetime of RNAs can be highly variable, and RNA degradation is tightly regulated. In bacteria, endonucleases generate mRNA fragments for destruction by exonucleases. In eukaryotes, mRNAs typically must be decapped and the poly(A) tail shortened before degradation. The exosome is a supramolecular complex of exo- and endonucleases involved in many steps of eukaryotic RNA decay.
 Many of the RNA molecules in bacteria and virtually all RNA molecules in eukaryotes are processed to some degree after synthesis. Processing can include addition or deletion of nucleotide sequences as well as chemical modification of RNA nucleotides. All of these events can be used to control the posttranscriptional fate of the RNA in the cell. As a result, many mature RNAs are not exact copies of the DNA genes from which they were transcribed. Some of the most interesting molecular events in RNA metabolism occur during posttranscriptional processing. Intriguingly, several of the enzymes that catalyze these reactions have active sites composed of RNA rather than protein. The discovery of these catalytic RNAs, or
Many of the RNA molecules in bacteria and virtually all RNA molecules in eukaryotes are processed to some degree after synthesis. Processing can include addition or deletion of nucleotide sequences as well as chemical modification of RNA nucleotides. All of these events can be used to control the posttranscriptional fate of the RNA in the cell. As a result, many mature RNAs are not exact copies of the DNA genes from which they were transcribed. Some of the most interesting molecular events in RNA metabolism occur during posttranscriptional processing. Intriguingly, several of the enzymes that catalyze these reactions have active sites composed of RNA rather than protein. The discovery of these catalytic RNAs, or 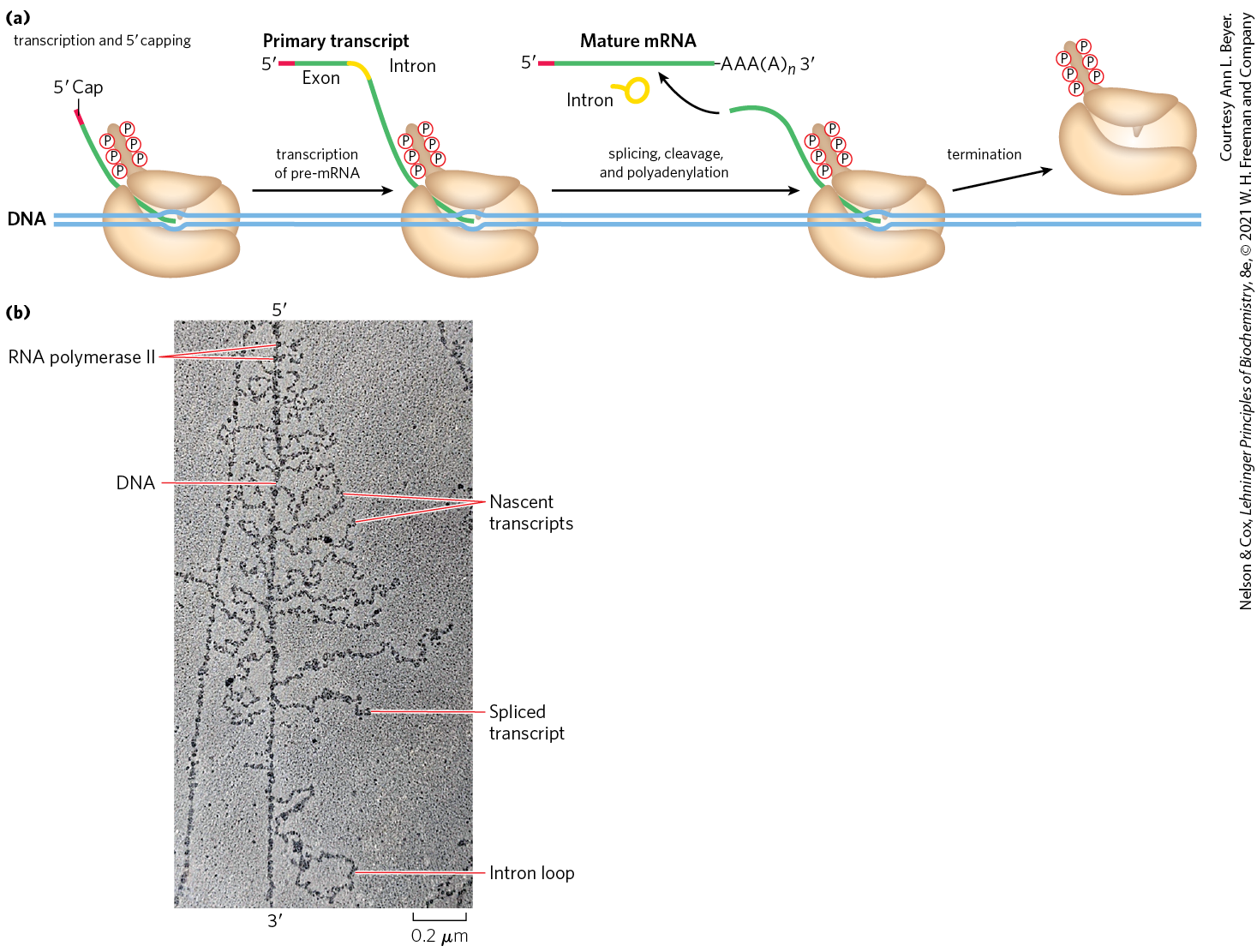


 through
through  ).
).
 The polyadenylate polymerase synthesizes a poly(A) tail 80 to 250 nucleotides long, beginning at the cleavage site.
The polyadenylate polymerase synthesizes a poly(A) tail 80 to 250 nucleotides long, beginning at the cleavage site.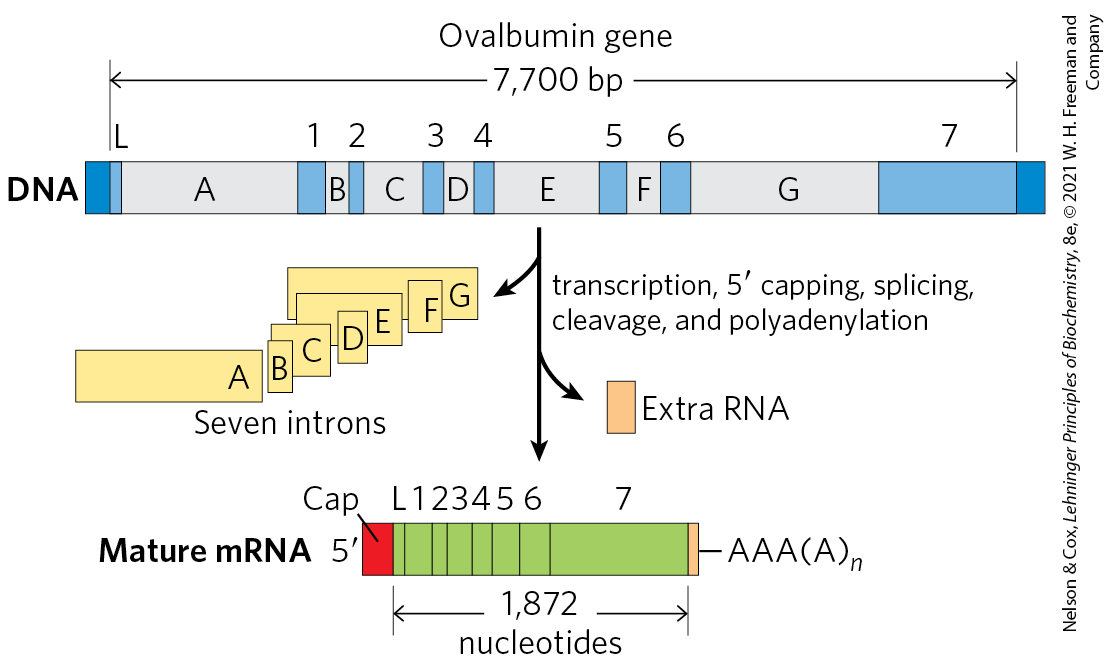

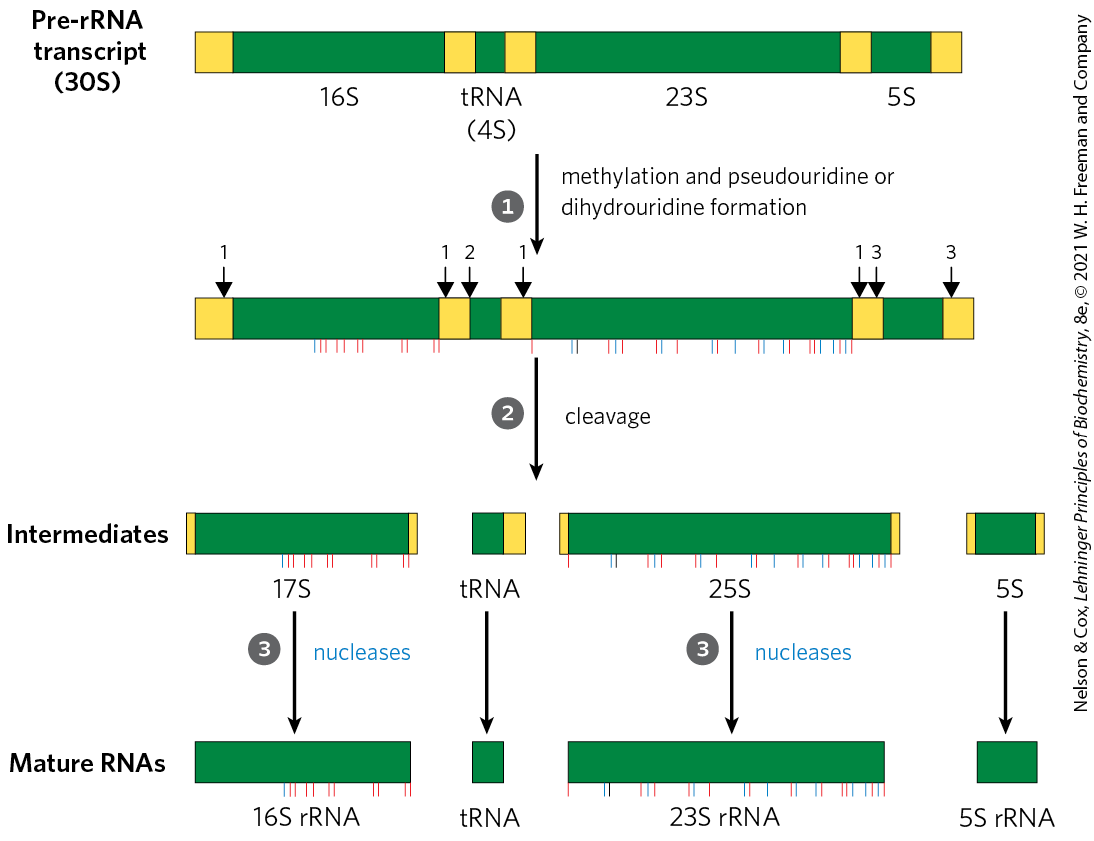
 The final 16S, 23S, and 5S rRNA products result from the action of a variety of specific nucleases. The seven copies of pre-rRNA gene in the E. coli chromosome differ in the number, location, and identity of tRNAs included in the primary transcript. Some copies of the gene have additional tRNA gene segments between the 16S and 23S rRNA segments and at the far end of the primary transcript.
The final 16S, 23S, and 5S rRNA products result from the action of a variety of specific nucleases. The seven copies of pre-rRNA gene in the E. coli chromosome differ in the number, location, and identity of tRNAs included in the primary transcript. Some copies of the gene have additional tRNA gene segments between the 16S and 23S rRNA segments and at the far end of the primary transcript.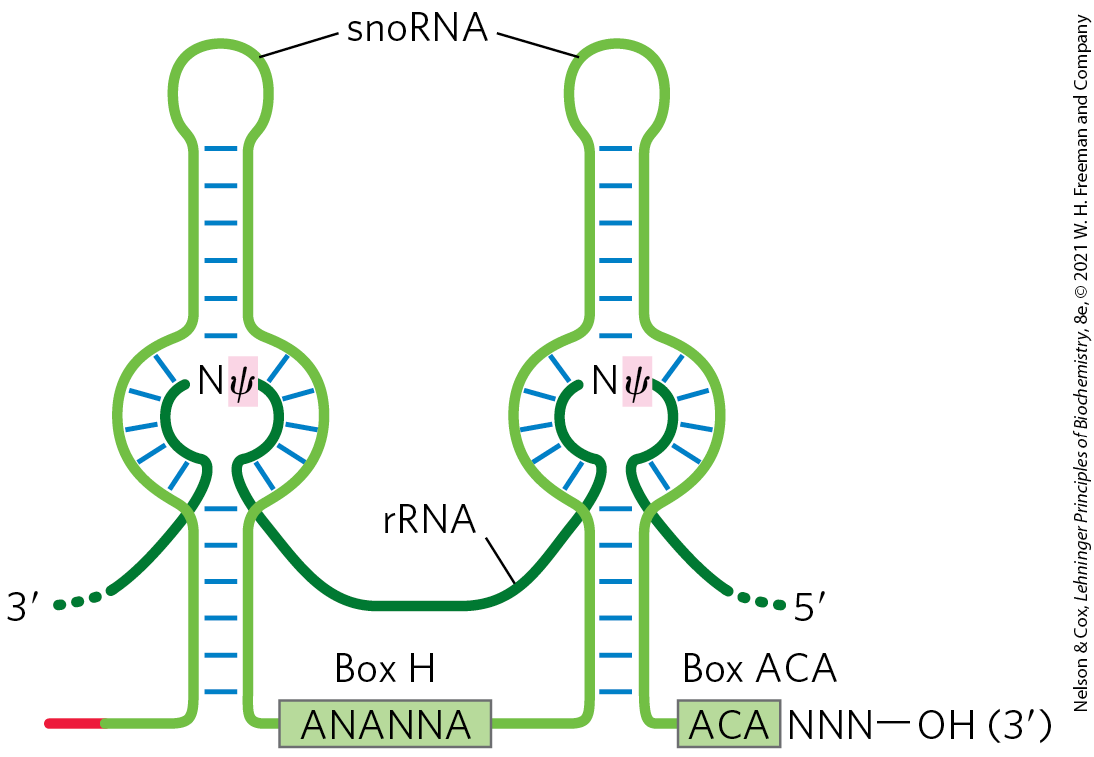
 Many primary transcripts produced in bacteria and eukaryotes must be processed into a mature form to be functional. Processing can include modifications to the and ends of the RNA, removal of internal RNA sequences by splicing, and modifications of the RNA nucleotides.
Many primary transcripts produced in bacteria and eukaryotes must be processed into a mature form to be functional. Processing can include modifications to the and ends of the RNA, removal of internal RNA sequences by splicing, and modifications of the RNA nucleotides.