19.5 Mitochondrial Genes: Their Origin and the Effects of Mutations
Mitochondria contain their own genome, a circular, double-stranded DNA (mtDNA) molecule. Each of the hundreds or thousands of mitochondria in a typical cell has about five copies of this genome. The human mitochondrial chromosome (Fig. 19-40) contains 37 genes (16,569 bp), including 13 that encode subunits of proteins of the respiratory chain (Table 19-6); the remaining genes code for rRNA and tRNA molecules essential to the protein-synthesizing machinery of mitochondria. To synthesize these 13 protein subunits, mitochondria have their own ribosomes, distinctly different from those in the cytoplasm. The great majority of mitochondrial proteins — about 1,200 different types — are encoded by nuclear genes, synthesized on cytoplasmic ribosomes, then imported into and assembled in the mitochondria (Chapter 27).
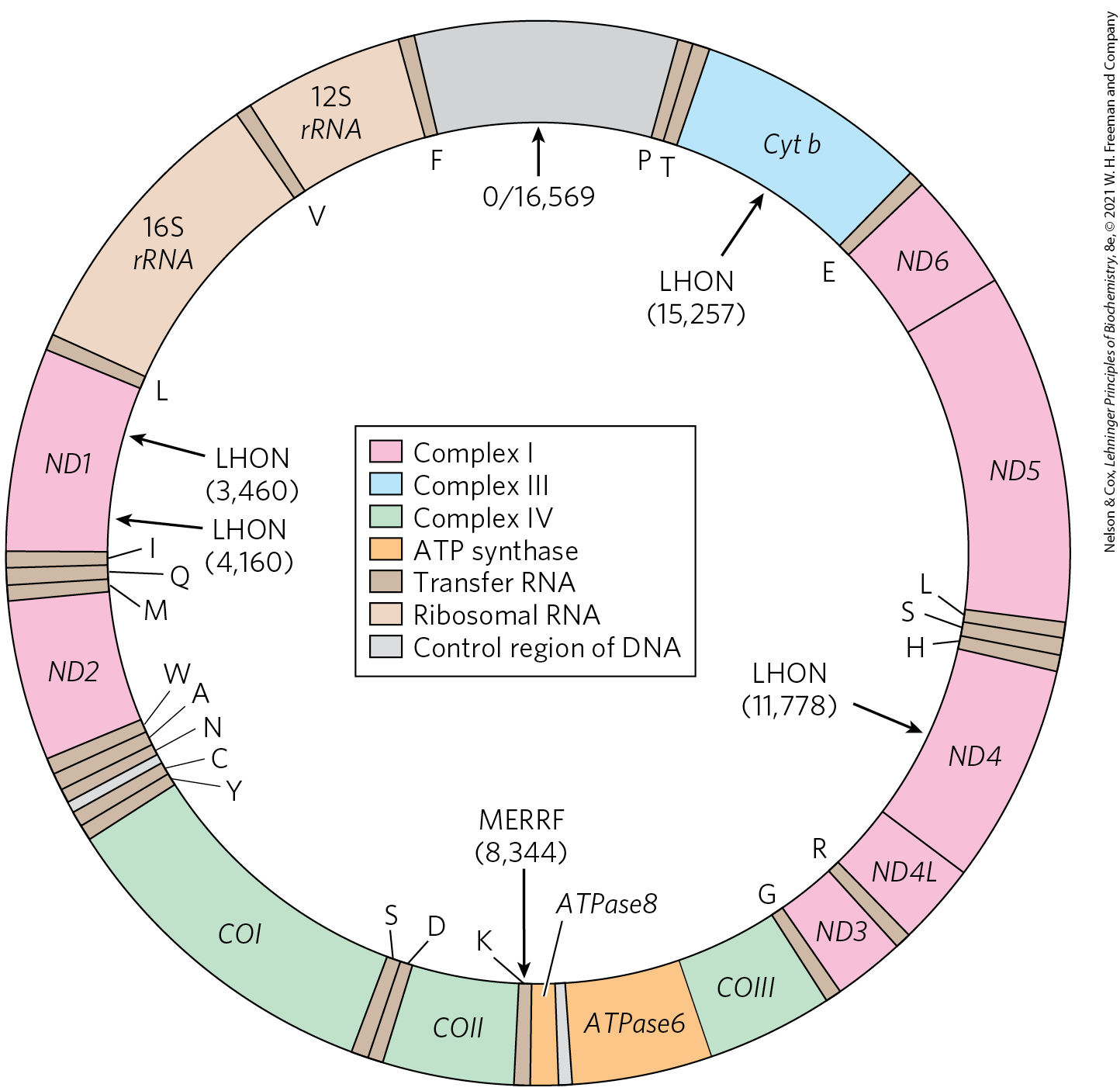
FIGURE 19-40 Mitochondrial genes and mutations. A map of human mitochondrial DNA, showing the genes that encode proteins of Complex I, the NADH dehydrogenase (ND1 to ND6); the cytochrome b of Complex III (Cyt b); the subunits of cytochrome oxidase, Complex IV (COI to COIII); and two subunits of ATP synthase (ATPase6 and ATPase8). The colors of the genes correspond to those of the complexes shown in Figure 19-7. Also included here are the genes for ribosomal RNAs (rRNA) and for some mitochondrion-specific transfer RNAs; tRNA specificity is indicated by the one-letter codes for amino acids. Arrows indicate the positions of mutations that cause Leber hereditary optic neuropathy (LHON) and myoclonic epilepsy with ragged-red fibers (MERRF) syndrome. Numbers in parentheses indicate the position of the altered nucleotides (nucleotide 1 is at the top of the circle, and numbering proceeds counterclockwise). [Information from M. A. Morris, J. Clin. Neuroophthalmol. 10:159, 1990.]
Complex |
Number of subunits |
Number of subunits encoded by mtDNA |
|---|---|---|
I NADH dehydrogenase |
45 |
7 |
II Succinate dehydrogenase |
4 |
0 |
III Ubiquinone:cytochrome c oxidoreductase |
11 |
1 |
IV Cytochrome oxidase |
13 |
3 |
V ATP synthase |
8 |
2 |
Mitochondria Evolved from Endosymbiotic Bacteria
The existence of mitochondrial DNA, ribosomes, and tRNAs supports the theory of the endosymbiotic origin of mitochondria (see Fig. 1-37), which holds that the first organisms capable of aerobic metabolism, including respiration-linked ATP production, were bacteria. Primitive eukaryotes that lived anaerobically (by fermentation) acquired the ability to carry out oxidative phosphorylation when they established a symbiotic relationship with bacteria living in their cytosol. After a long period of evolution and the movement of many bacterial genes into the nucleus of the “host” eukaryote, the endosymbiotic bacteria eventually became mitochondria.
 Primitive eukaryotes that lived anaerobically (by fermentation) acquired the ability to carry out oxidative phosphorylation when they established a symbiotic relationship with bacteria living in their cytosol. After a long period of evolution and the movement of many bacterial genes into the nucleus of the “host” eukaryote, the endosymbiotic bacteria eventually became mitochondria.
Primitive eukaryotes that lived anaerobically (by fermentation) acquired the ability to carry out oxidative phosphorylation when they established a symbiotic relationship with bacteria living in their cytosol. After a long period of evolution and the movement of many bacterial genes into the nucleus of the “host” eukaryote, the endosymbiotic bacteria eventually became mitochondria.This hypothesis presumes that early free-living bacteria had the enzymatic machinery for oxidative phosphorylation. And it predicts that their modern bacterial descendants must have respiratory chains closely similar to those of modern eukaryotes. They do. Aerobic bacteria carry out NAD-linked electron transfer from substrates to , coupled to the phosphorylation of cytosolic ADP. The dehydrogenases are located in the bacterial cytosol, and the respiratory chain in the plasma membrane. The electron carriers translocate protons outward across the plasma membrane as electrons are transferred to . Bacteria such as E. coli have complexes in their plasma membranes; the portion protrudes into the cytosol and catalyzes ATP synthesis from ADP and as protons flow back into the cell through the proton channel of .
The respiration-linked extrusion of protons across the bacterial plasma membrane also provides the driving force for other processes. Certain bacterial transport systems bring about uptake of extracellular nutrients (lactose, for example) against a concentration gradient, in symport with protons. And the rotary motion of bacterial flagella is provided by “proton turbines,” molecular rotary motors driven not by ATP but directly by the transmembrane electrochemical potential generated by respiration-linked proton pumping (Fig. 19-41). It seems likely that the chemiosmotic mechanism evolved early, before the emergence of eukaryotes.
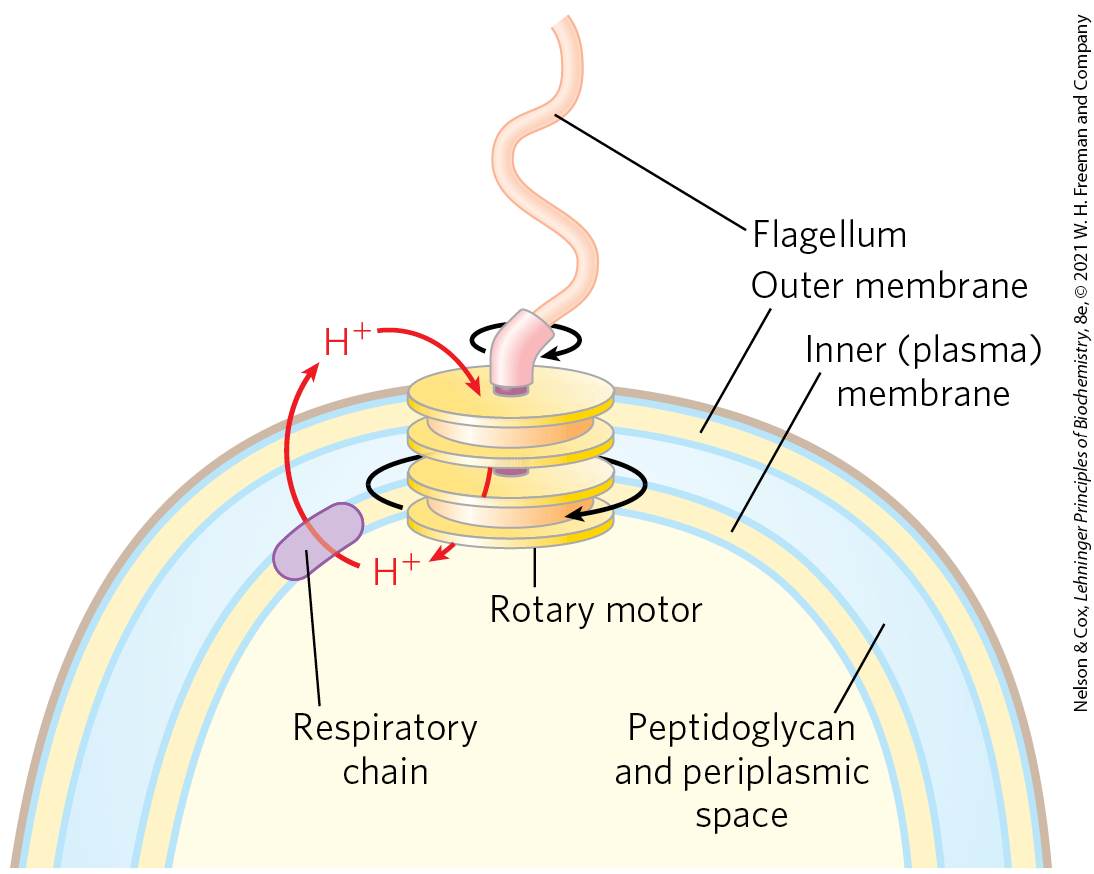
FIGURE 19-41 Rotation of bacterial flagella by proton-motive force. The shaft and rings at the base of the flagellum make up a rotary motor that has been called a “proton turbine.” Protons ejected by electron transfer flow back into the cell through the turbine, causing rotation of the shaft of the flagellum. This motion differs fundamentally from the motion of muscle and of eukaryotic flagella and cilia, for which ATP hydrolysis is the energy source.
Mutations in Mitochondrial DNA Accumulate throughout the Life of the Organism
The respiratory chain is the major producer of reactive oxygen species in cells, so mitochondrial contents, including the mitochondrial genome, suffer the greatest exposure to, and damage by, ROS. Moreover, the mitochondrial DNA replication system is less effective than the nuclear system at correcting mistakes made during replication and at repairing DNA damage. As a consequence, defects in mtDNA accumulate over time. One theory of aging is that this gradual accumulation of defects is the primary cause of many of the “symptoms” of aging, which include, for example, progressive weakening of skeletal and heart muscle.
A unique feature of mitochondrial inheritance is the variation among individual cells, and between one individual organism and another, in the effects of a mtDNA mutation. A typical cell has hundreds or thousands of mitochondria, each with multiple copies of its own genome (Fig. 19-2b). Animals inherit essentially all of their mitochondria from the female parent. Eggs are large and contain or mitochondria; sperm are much smaller and contain far fewer mitochondria — perhaps 100 to 1,000. Furthermore, there is an active mechanism for targeting sperm-derived mitochondria for degradation in the fertilized egg. Just after fertilization, maternal phagosomes migrate to the site of sperm entry, engulf sperm mitochondria, and degrade them.
Suppose that, in a female organism, damage to one mitochondrial genome occurs in a germ cell from which oocytes develop, such that the germ cell contains mainly mitochondria with wild-type genes but one mitochondrion with a mutant gene. During the course of oocyte maturation, as this germ cell and its descendants repeatedly divide, the defective mitochondrion replicates and its progeny, all defective, are randomly distributed to daughter cells. Eventually, the mature egg cells contain different proportions of the defective mitochondria. When an egg cell is fertilized and undergoes the many divisions of embryonic development, the resulting somatic cells differ in their proportion of mutant mitochondria (Fig. 19-42a). This heteroplasmy (in contrast to homoplasmy, in which every mitochondrial genome in every cell is the same) results in mutant phenotypes of varying degrees of severity. Cells (and tissues) containing mostly wild-type mitochondria have the wild-type phenotype; they are essentially normal. Other heteroplasmic cells have intermediate phenotypes, some almost normal, others (with a high proportion of mutant mitochondria) abnormal (Fig. 19-42b). If the abnormal phenotype is associated with a disease, individuals with the same mtDNA mutation may have disease symptoms of differing severity — depending on the number and distribution of affected mitochondria.
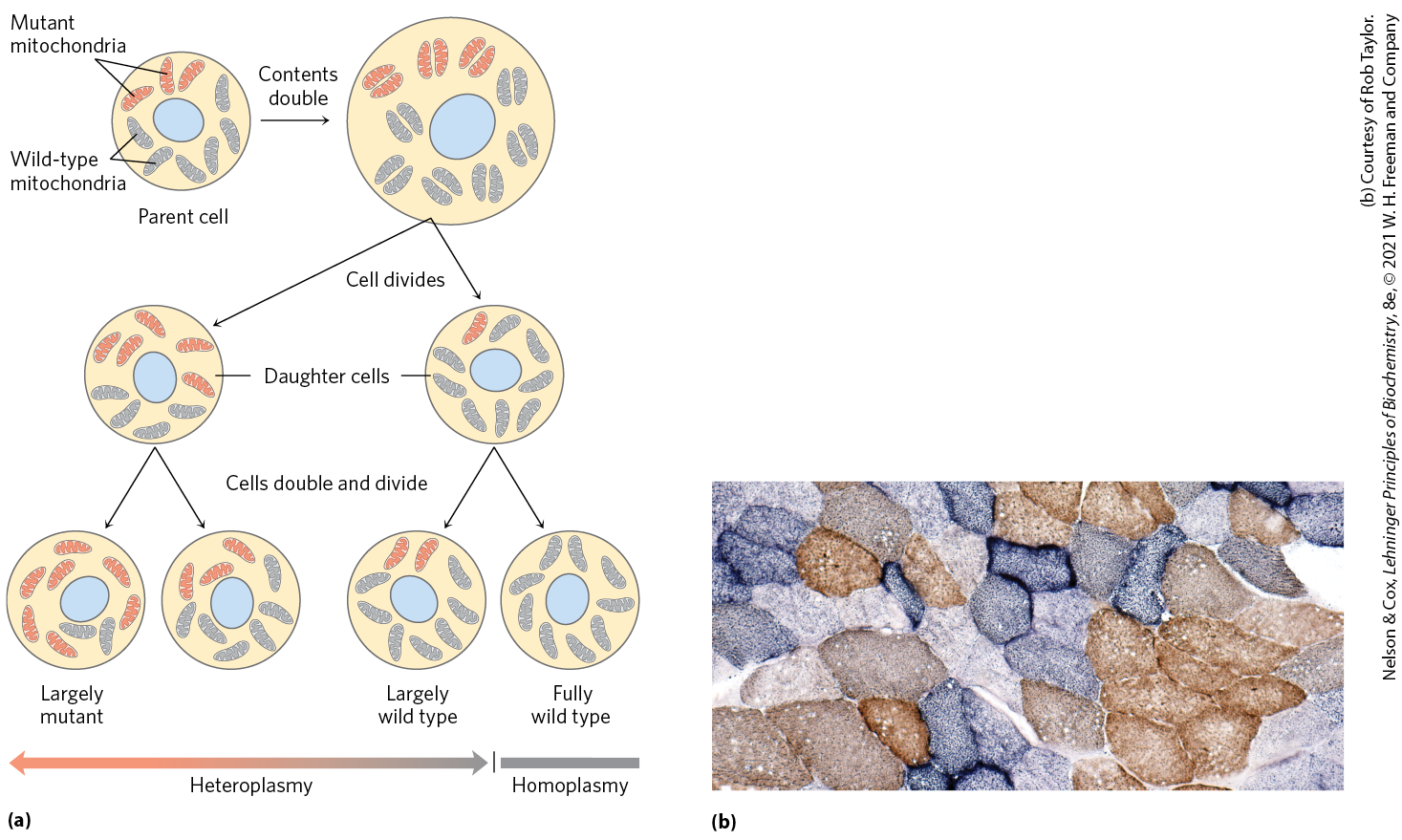
FIGURE 19-42 Heteroplasmy in mitochondrial genomes. (a) When a mature egg cell is fertilized, all of the mitochondria in the resulting diploid cell (zygote) are maternal; none come from the sperm. If some fraction of the maternal mitochondria have a mutant gene, the random distribution of mitochondria during subsequent cell divisions yields some daughter cells with mostly mutant mitochondria, some with mostly wild-type mitochondria, and some in between. Thus daughter cells show a varying degree of heteroplasmy. (b) Different degrees of heteroplasmy produce different cellular phenotypes. This section of human muscle tissue is from an individual with defective cytochrome oxidase. The cells were stained so that wild-type cells are blue and cells with mutant cytochrome oxidase are brown. As the micrograph shows, different cells in the same tissue are affected to different degrees by the mitochondrial mutation. [(b) Courtesy of Rob Taylor. Reprinted with permission from R. W. Taylor and D. M. Turnbull, Nat. Rev. Genet. 6:389, 2005, Fig. 2a.]
Some Mutations in Mitochondrial Genomes Cause Disease
About 1 in 5,000 people have a disease-causing mutation in a mitochondrial protein that reduces the cell’s capacity to produce ATP. A growing number of these diseases have been attributed to mutations in mitochondrial genes. Some tissues and cell types — neurons, myocytes of both skeletal and cardiac muscle, and β cells of the pancreas — are less able than others to tolerate lowered ATP production and are therefore more affected by mutations in mitochondrial proteins.
 About 1 in 5,000 people have a disease-causing mutation in a mitochondrial protein that reduces the cell’s capacity to produce ATP. A growing number of these diseases have been attributed to mutations in mitochondrial genes. Some tissues and cell types — neurons, myocytes of both skeletal and cardiac muscle, and β cells of the pancreas — are less able than others to tolerate lowered ATP production and are therefore more affected by mutations in mitochondrial proteins.
About 1 in 5,000 people have a disease-causing mutation in a mitochondrial protein that reduces the cell’s capacity to produce ATP. A growing number of these diseases have been attributed to mutations in mitochondrial genes. Some tissues and cell types — neurons, myocytes of both skeletal and cardiac muscle, and β cells of the pancreas — are less able than others to tolerate lowered ATP production and are therefore more affected by mutations in mitochondrial proteins.A group of genetic diseases known as the mitochondrial encephalomyopathies affect primarily the brain and skeletal muscle. These diseases are invariably inherited from the mother, because, as noted above, a developing embryo derives all its mitochondria from the egg. The rare disease Leber hereditary optic neuropathy (LHON) affects the central nervous system, including the optic nerves, causing bilateral loss of vision in early adulthood. A single base change in the mitochondrial gene ND4 (Fig. 19-40) changes an Arg residue to a His residue in a polypeptide of Complex I, and the result is mitochondria partially defective in electron transfer from NADH to ubiquinone. Although these mitochondria can produce some ATP by electron transfer from succinate, they apparently cannot supply sufficient ATP to support the very active metabolism of neurons, including the optic nerve. A single base change in the mitochondrial gene for cytochrome b, a component of Complex III, also produces LHON, demonstrating that the pathology results from a general reduction of mitochondrial function, not specifically from a defect in electron transfer through Complex I.
A mutation in the mitochondrial gene ATP6 affects the proton pore in ATP synthase, leading to low rates of ATP synthesis while leaving the respiratory chain intact. Oxidative stress due to the continued supply of electrons from NADH increases the production of ROS, and the damage to mitochondria caused by ROS sets up a vicious cycle. Half of individuals with this mutant gene die within days or months of birth.
Myoclonic epilepsy with ragged-red fibers (MERRF) syndrome is caused by a mutation in the mitochondrial gene that encodes a tRNA specific for lysine . This disease, characterized by uncontrollable muscular jerking, results from defective production of several of the proteins that require mitochondrial tRNAs for their synthesis. Skeletal muscle fibers of individuals with MERRF syndrome have abnormally shaped mitochondria that sometimes contain paracrystalline structures (Fig. 19-43). Other mutations in mitochondrial genes are believed to be responsible for the progressive muscular weakness that characterizes mitochondrial myopathy and for enlargement and deterioration of the heart muscle in hypertrophic cardiomyopathy.
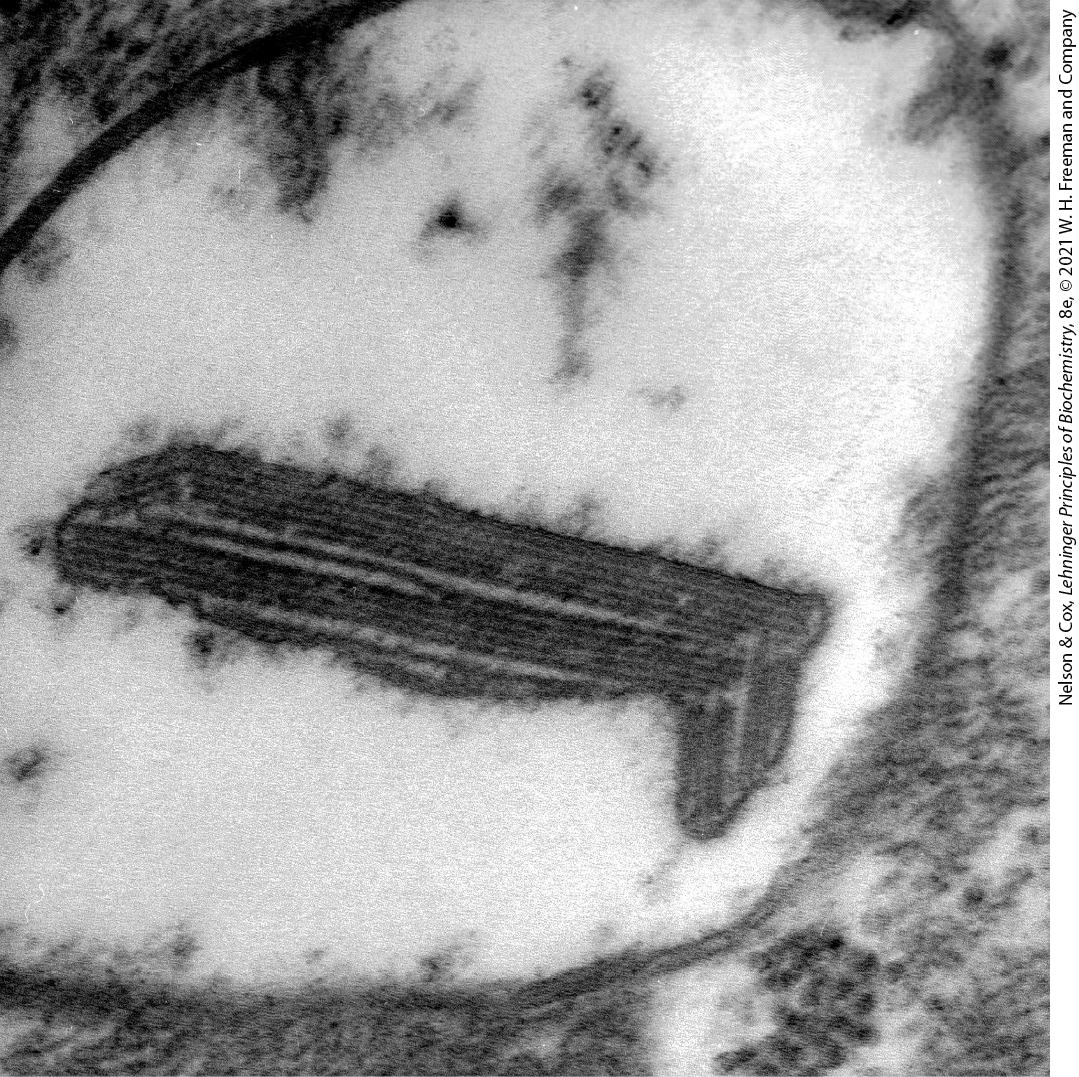
FIGURE 19-43 Paracrystalline inclusions in MERRF syndrome mitochondrion. Electron micrograph of an abnormal mitochondrion from the muscle of an individual with MERRF syndrome, showing the paracrystalline protein inclusions sometimes present in the mutant mitochondria. [From Regionalized Pathology Correlates with Augmentation of mtDNA Copy Numbers in a Patient with Myoclonic Epilepsy with Ragged-Red Fibers (MERRF-Syndrome). PLOS ONE, Anja Brinckmann et al., October 20, 2010. https://doi.org/10.1371/journal.pone.0013513]
If a prospective mother is known to carry a pathogenic mitochondrial gene, the technique of mitochondrial donation can circumvent the passage of that mutant gene to her offspring. A prospective mother’s nuclear genes are microscopically transplanted into an enucleated ovum from a donor with healthy mitochondria, then the ovum is fertilized in vitro and the resulting embryo is transplanted into the mother’s uterus. This and similar “three-parent baby” procedures, which were approved in the United Kingdom in 2015, raise ethical issues that are being vigorously debated.
Mitochondrial disease can also result from mutations in any of the ~1,200 nuclear genes that encode mitochondrial proteins. For example, a mutation in one of the nuclear-encoded proteins of Complex IV, COX6B1, results in severe defects in brain development and thickened walls of the heart muscle. Other nuclear genes encode proteins essential for the assembly of mitochondrial complexes. Mutations in these genes can also lead to serious mitochondrial disease.

A Rare Form of Diabetes Results from Defects in the Mitochondria of Pancreatic β Cells
The insulin so important to glucose homeostasis in all humans is produced and exported from pancreatic β cells. Insulin export hinges on the ATP concentration in those cells. When blood glucose is high, β cells take up glucose and oxidize it by glycolysis and the citric acid cycle, raising [ATP] above a threshold level (Fig. 19-44). When [ATP] exceeds this threshold, an ATP-gated channel in the plasma membrane closes, depolarizing the membrane and triggering insulin release (see Fig. 23-24).
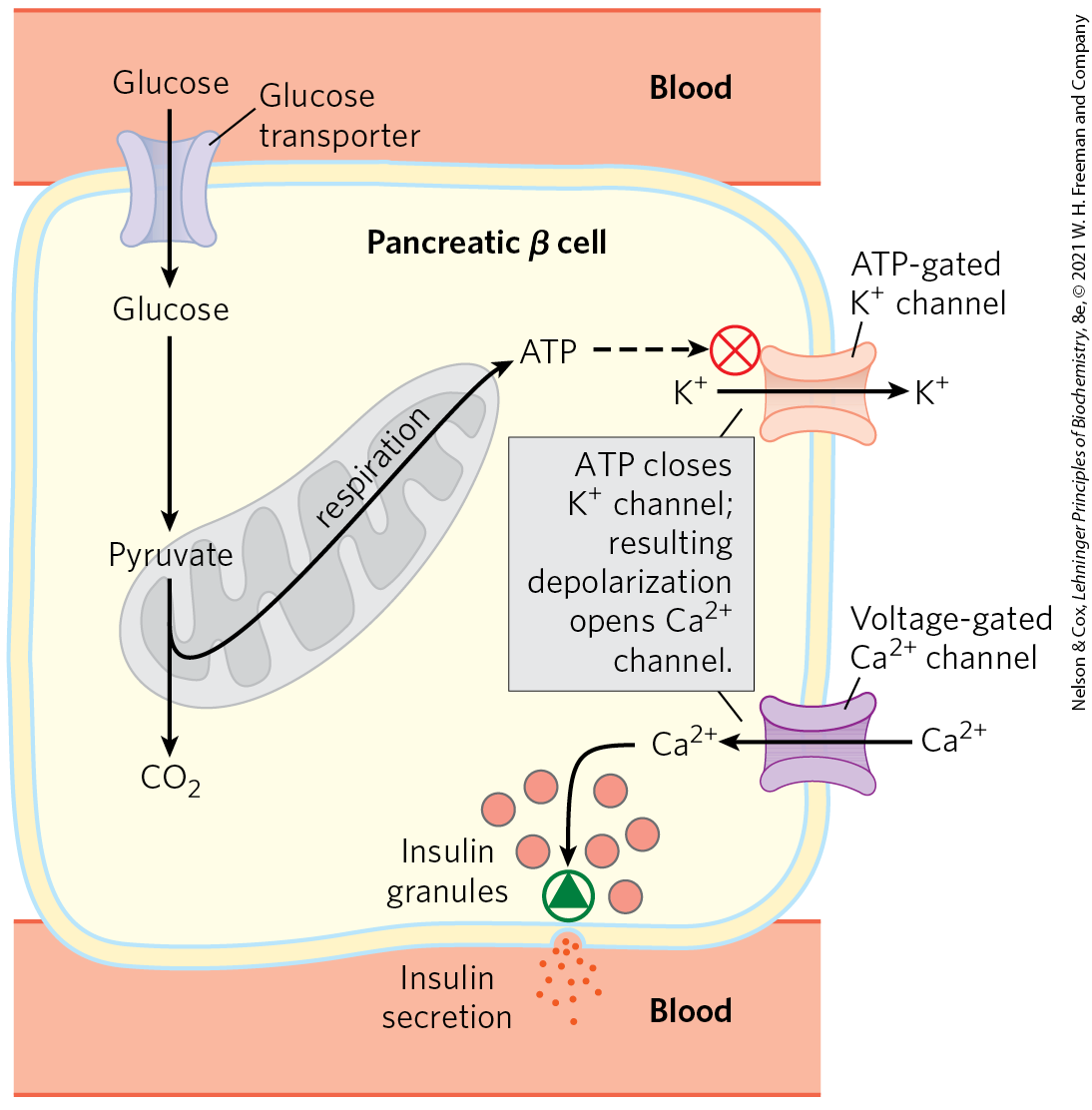
FIGURE 19-44 A mitochondrial defect prevents insulin secretion. In the normal situation, as depicted here, when the blood glucose level rises, production of ATP in β cells increases. ATP, by blocking channels, depolarizes the plasma membrane and thus opens voltage-gated channels. The resulting influx of triggers exocytosis of insulin-containing secretory vesicles, releasing insulin. When oxidative phosphorylation in β cells is defective, [ATP] is never sufficient to trigger this process, and insulin is not released.
Normal insulin release can be compromised in several ways. Pancreatic β cells with defects in any aspect of oxidative phosphorylation may not be able to increase [ATP] above this threshold, and the resulting failure of insulin release effectively produces diabetes. For example, defects in the gene for glucokinase, the hexokinase IV isozyme present in β cells, lead to a rare form of diabetes called MODY2 (maturity onset diabetes of the young); low glucokinase activity prevents the generation of ATP concentrations above the threshold needed to trigger insulin secretion. Mutations in the mitochondrial or genes also compromise mitochondrial ATP production by limiting the expression of electron transfer components encoded in the mitochondrial DNA. Type 2 diabetes mellitus is common among individuals with these defects (although such cases make up a very small fraction of all cases of diabetes).
When nicotinamide nucleotide transhydrogenase, which is part of the mitochondrial defense against ROS (Fig. 19-18), is genetically defective, the accumulation of ROS damages mitochondria, slowing ATP production and blocking insulin release by β cells (Fig. 19-44). Damage caused by ROS, including damage to mtDNA, may also underlie other human diseases; there is some evidence for its involvement in Alzheimer, Parkinson, and Huntington diseases and in heart failure, as well as in aging.
SUMMARY 19.5 Mitochondrial Genes: Their Origin and the Effects of Mutations
- A small proportion of human mitochondrial proteins, 13 in all, are encoded by the mitochondrial genome and synthesized in mitochondria. About 1,200 mitochondrial proteins are encoded by nuclear genes and imported into mitochondria after their synthesis.
- Mitochondria arose from aerobic bacteria that entered into an endosymbiotic relationship with ancestral eukaryotes.
- Mutations in the mitochondrial genome accumulate over the life of the organism. Mutations in nuclear or mitochondrial genes that encode components of the respiratory chain, ATP synthase, and the ROS-scavenging system, and even in tRNA genes, can cause a variety of human diseases, which often most severely affect muscle, heart, pancreatic β cells, and brain.
- It is possible to combine the mitochondria from one woman with the nuclear genes of another to create an ovum free of a mutation that would have led to a mitochondrial disease.
- Mitochondrial defects in pancreatic β cells that limit ATP production when glucose levels are high can compromise normal insulin release and give rise to a form of type 2 diabetes.
 A small proportion of human mitochondrial proteins, 13 in all, are encoded by the mitochondrial genome and synthesized in mitochondria. About 1,200 mitochondrial proteins are encoded by nuclear genes and imported into mitochondria after their synthesis.
A small proportion of human mitochondrial proteins, 13 in all, are encoded by the mitochondrial genome and synthesized in mitochondria. About 1,200 mitochondrial proteins are encoded by nuclear genes and imported into mitochondria after their synthesis.