15.3 Coordinated Regulation of Glycogen Breakdown and Synthesis
As we have seen, the mobilization of stored glycogen is brought about by glycogen phosphorylase, which degrades glycogen to glucose 1-phosphate (Fig. 15-3). Glycogen phosphorylase provides an especially instructive case of enzyme regulation. It was one of the first known examples of an allosterically regulated enzyme and the first enzyme shown to be controlled by reversible phosphorylation. It was also one of the first allosteric enzymes for which the detailed three-dimensional structures of the active and inactive forms were revealed by x-ray crystallographic studies. Glycogen phosphorylase also illustrates how isozymes play their tissue-specific roles.
Glycogen Phosphorylase Is Regulated by Hormone-Stimulated Phosphorylation and by Allosteric Effectors

Earl W. Sutherland, Jr., 1915–1974
In the late 1930s, Carl and Gerty Cori (Box 15-1) discovered that the glycogen phosphorylase of skeletal muscle exists in two interconvertible forms: glycogen phosphorylase a, which is catalytically active, and glycogen phosphorylase b, which is much less active. (Note that glycogen phosphorylase is often referred to simply as phosphorylase — so honored because it was the first phosphorylase to be discovered; the shortened name has persisted in common usage and in the literature.) Subsequent studies by Earl Sutherland showed that phosphorylase b predominates in resting muscle, but during vigorous muscular activity, epinephrine triggers phosphorylation of phosphorylase b, converting it to its more active form, phosphorylase a (Fig. 15-11). In the liver, glucagon triggers phosphorylation of phosphorylase b, converting it to its active form.
 In the late 1930s, Carl and Gerty Cori (
In the late 1930s, Carl and Gerty Cori (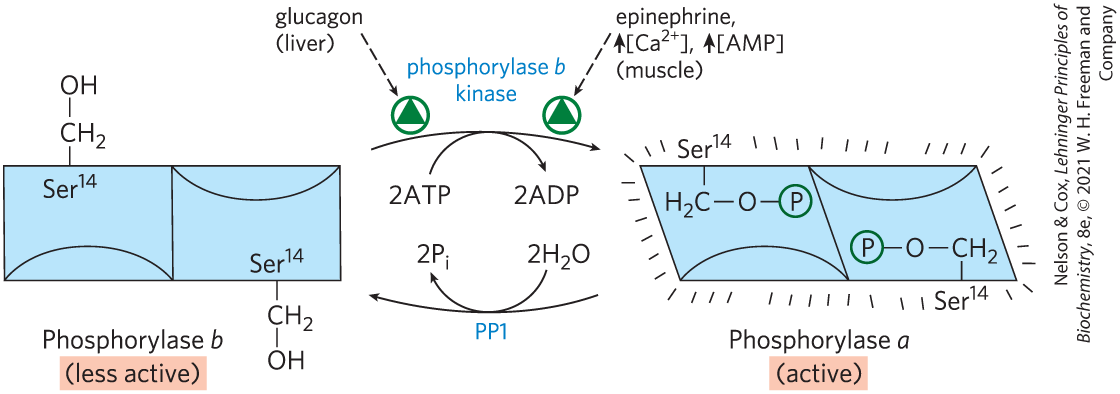
FIGURE 15-11 Regulation of muscle glycogen phosphorylase by covalent modification. In the more active form of the enzyme, phosphorylase a, residues, one on each subunit, are phosphorylated. Phosphorylase a is converted to the less active form, phosphorylase b, by enzymatic loss of these phosphoryl groups, catalyzed by phosphoprotein phosphatase 1 (PP1). Phosphorylase b can be reconverted (reactivated) to phosphorylase a by the action of phosphorylase b kinase.
Sutherland discovered the second messenger cAMP, which increases in concentration in response to stimulation by epinephrine (in muscle; see Fig. 12-4) or glucagon (in liver). Elevated [cAMP] initiates an enzyme cascade (Fig. 15-12), in which a catalyst activates a catalyst, which activates a catalyst. As we saw in Chapter 12, such cascades allow exponential amplification of the initial signal. The rise in [cAMP] activates cAMP-dependent protein kinase, also called protein kinase A (PKA). PKA then phosphorylates and activates phosphorylase b kinase, which catalyzes the phosphorylation of glycogen phosphorylase b, activating it and thus stimulating glycogen breakdown.
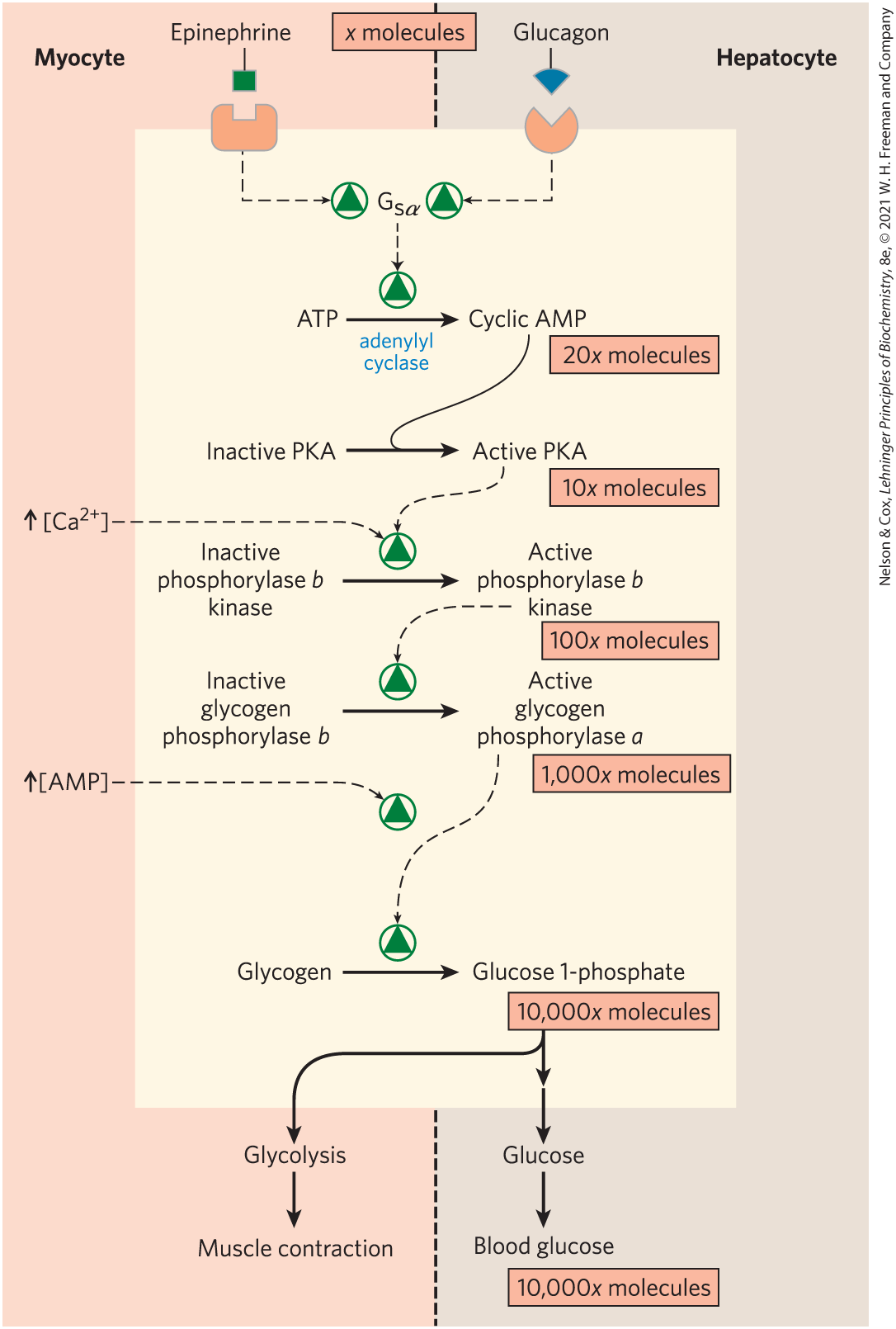
FIGURE 15-12 Cascade mechanism of epinephrine and glucagon action. By binding to specific surface receptors, either epinephrine acting on a myocyte (left) or glucagon acting on a hepatocyte (right) activates a GTP-binding protein, . Active triggers a rise in [cAMP], activating PKA. This sets off a cascade of phosphorylations; PKA activates phosphorylase b kinase, which then activates glycogen phosphorylase. Such cascades effect a large amplification of the initial signal; the figures in pink boxes are certainly low estimates of the actual increase in number of molecules at each stage of the cascade. The resulting breakdown of glycogen provides glucose, which in the myocyte can supply ATP (via glycolysis) for muscle contraction and in the hepatocyte is released into the blood to counter the low blood glucose.
In muscle, this provides fuel for glycolysis to sustain muscle contraction for the fight-or-flight response signaled by epinephrine. In liver, glycogen breakdown counters the low blood glucose signaled by glucagon, by releasing glucose into the blood. These different roles are reflected in subtle differences in the regulatory mechanisms in muscle and liver. The glycogen phosphorylases of liver and muscle are isozymes, encoded by different genes and differing in their regulatory properties.
Figure 15-12 shows the pathway for hormonal regulation of glycogen phosphorylase activity: epinephrine or glucagon initiates the cascade that activates phosphorylase b kinase. Phosphorylase b kinase activates phosphorylase by transferring a phosphoryl group to on each of the two identical subunits of phosphorylase b, triggering a conformation change from the T state (phosphorylase b) to the R state (phosphorylase a), shown in detail in Figure 6-40. Superimposed on the hormonal activation of phosphorylase b kinase are allosteric control mechanisms (Fig. 15-12). In muscle, , the signal for muscle contraction, binds to and activates phosphorylase b kinase. binds to phosphorylase b kinase through its δ subunit, which is calmodulin (see Fig. 12-17). AMP, which accumulates in vigorously contracting muscle as a result of ATP breakdown, binds to and activates phosphorylase, speeding the release of glucose 1-phosphate from glycogen. When ATP levels are adequate, ATP blocks the allosteric site to which AMP binds (see Fig. 6-40), inactivating phosphorylase.
When the muscle returns to rest, a second enzyme, phosphoprotein phosphatase 1 (PP1), removes the phosphoryl groups from phosphorylase a, converting it to the less active form, phosphorylase b (see Fig. 15-11).
Like the enzyme of muscle, the glycogen phosphorylase of liver is regulated hormonally (by phosphorylation/dephosphorylation) and allosterically. The dephosphorylated form is essentially inactive. When the blood glucose level is too low, glucagon (acting through the cascade mechanism shown in Fig. 15-12) activates phosphorylase b kinase, which in turn converts phosphorylase b to its active a form, initiating the release of glucose into the blood. When blood glucose levels return to normal, glucose enters hepatocytes and binds to an inhibitory allosteric site on phosphorylase a. This binding also produces a conformational change that exposes the phosphorylated Ser residues to PP1, which catalyzes their dephosphorylation and inactivates the phosphorylase (Fig. 15-13). The allosteric site for glucose allows liver glycogen phosphorylase to act as its own glucose sensor and to respond appropriately to changes in blood glucose.
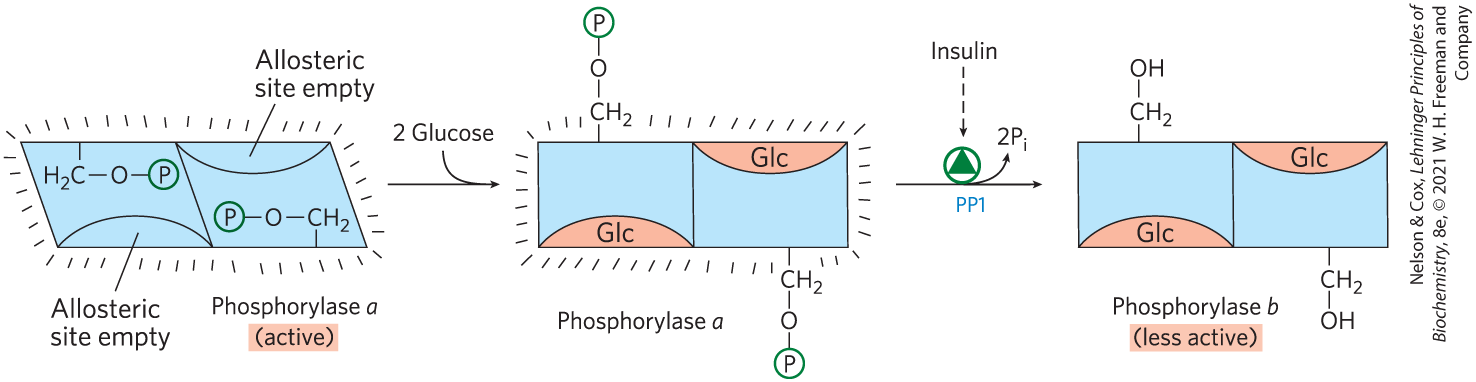
FIGURE 15-13 Glycogen phosphorylase of liver as a glucose sensor. Glucose binding to an allosteric site of the phosphorylase a isozyme of liver induces a conformational change that exposes its phosphorylated Ser residues to the action of phosphoprotein phosphatase 1 (PP1). This phosphatase converts phosphorylase a to phosphorylase b, sharply reducing the activity of phosphorylase and slowing glycogen breakdown in response to high blood glucose. Insulin also acts indirectly to stimulate PP1 and slow glycogen breakdown.
Glycogen Synthase Also Is Subject to Multiple Levels of Regulation
Like glycogen phosphorylase, glycogen synthase can exist in phosphorylated and unphosphorylated forms (Fig. 15-14). Its active form, glycogen synthase a, is unphosphorylated. Glycogen synthase kinase 3 (GSK3) adds phosphoryl groups to three Ser residues near the carboxyl terminus of glycogen synthase a converting it to glycogen synthase b, which is inactive unless its allosteric activator, glucose 6-phosphate, is present. The action of GSK3 is hierarchical; it cannot phosphorylate glycogen synthase until another protein kinase, casein kinase II (CKII), has first phosphorylated the glycogen synthase on a nearby residue, an event called priming (Fig. 15-15a). AMP-activated protein kinase (AMPK), which associates with glycogen granules through its carbohydrate-binding domain, also phosphorylates glycogen synthase, inhibiting glycogen synthesis during periods of metabolic stress, signaled by high [AMP] and low [ATP].
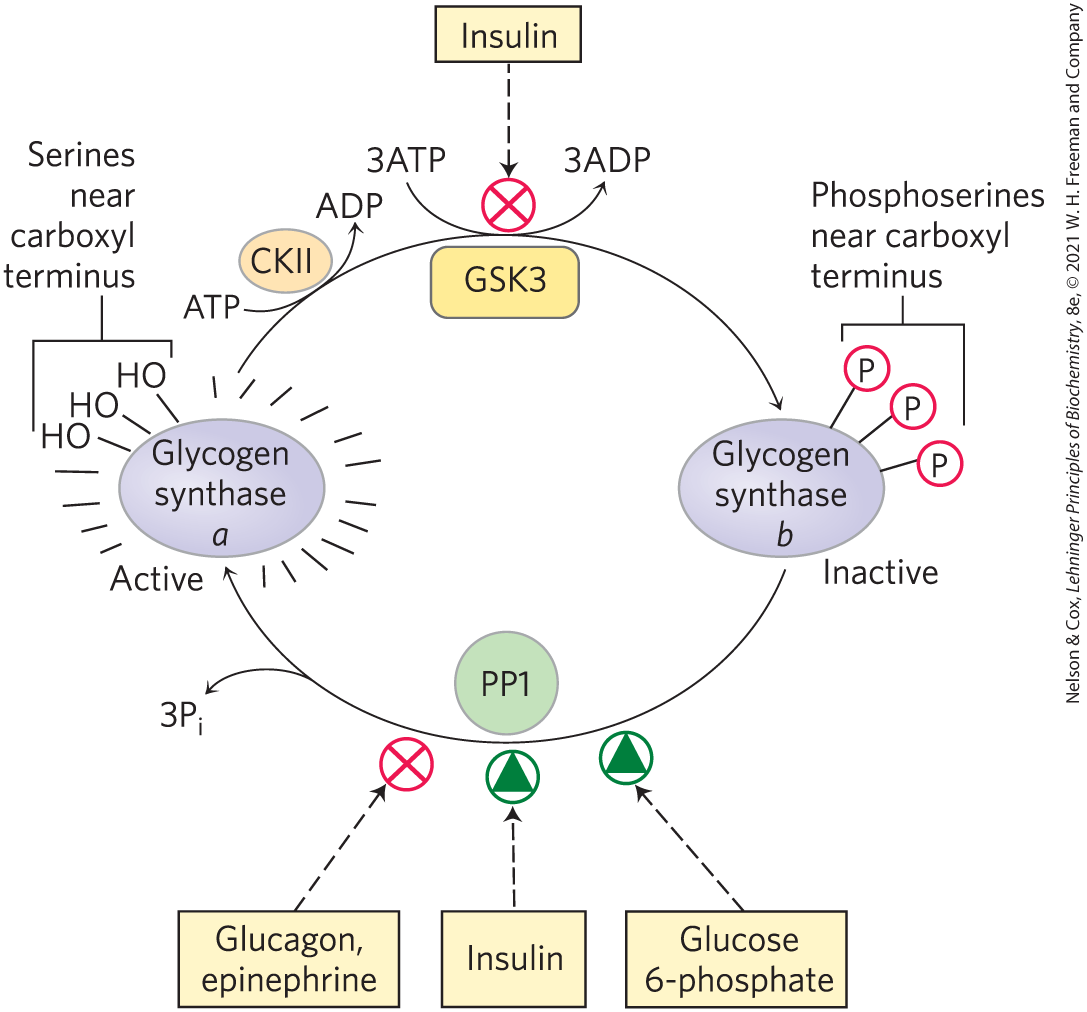
FIGURE 15-14 Effects of GSK3 on glycogen synthase activity. Glycogen synthase a, the active form, has three Ser residues near its carboxyl terminus. Their phosphorylation by glycogen synthase kinase 3 (GSK3) converts glycogen synthase to its inactive b form. Insulin favors the active a form of glycogen synthase by blocking the activity of GSK3 and activating phosphoprotein phosphatase 1 (PP1). In muscle, epinephrine activates PKA, which phosphorylates the glycogen-targeting protein Gm on a site that causes dissociation of PP1 from glycogen. Glucose 6-phosphate favors dephosphorylation of glycogen synthase by binding to it and promoting a conformation that is a good substrate for PP1.
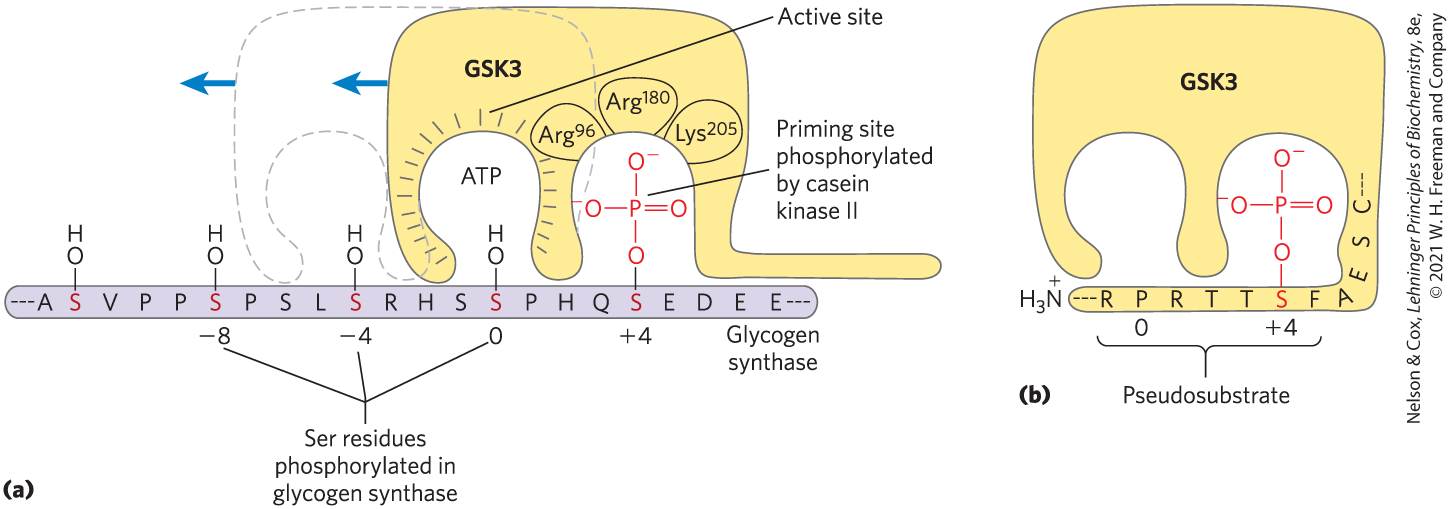
FIGURE 15-15 Priming of GSK3 phosphorylation of glycogen synthase. (a) Glycogen synthase kinase 3 first associates with its substrate (glycogen synthase) by interaction between three positively charged residues and a phosphoserine residue at position in the substrate. (For orientation, the Ser or Thr residue to be phosphorylated in the substrate is assigned the index 0. Residues on the amino-terminal side of this residue are numbered and so forth; residues on the carboxyl-terminal side are numbered and so forth.) This association aligns the active site of the enzyme with a Ser residue at position 0, which it phosphorylates. This creates a new priming site, and the enzyme moves down the protein to phosphorylate the Ser residue at position , and then the Ser at . (b) GSK3 has a Ser residue near its amino terminus that can be phosphorylated by PKA or PKB. This produces a “pseudosubstrate” region in GSK3 that folds into the priming site and makes the active site inaccessible to another protein substrate, inhibiting GSK3 until the priming phosphoryl group of its pseudosubstrate region is removed by PP1. Other proteins that are substrates for GSK3 also have a priming site at position , which must be phosphorylated by another protein kinase before GSK3 can act on them.
Insulin favors activation of glycogen synthase by blocking the activity of GSK3 and activating PP1. In muscle, epinephrine activates PKA, which phosphorylates the glycogen-targeting protein Gm (see Fig. 15-16), a regulatory subunit of PP1 in muscle, on a site that causes dissociation of PP1 from glycogen, effectively blocking its action on glycogen synthase.
In liver, conversion of glycogen synthase b to the active form is promoted by PP1, which is bound to the glycogen particle by its regulatory subunit in liver, Gl. PP1 removes the phosphoryl groups from the three Ser residues phosphorylated by GSK3. Glucose 6-phosphate binds to an allosteric site on glycogen synthase, making the enzyme a better substrate for dephosphorylation by PP1 and causing its activation. By analogy with glycogen phosphorylase, which acts as a glucose sensor, glycogen synthase can be regarded as a glucose 6-phosphate sensor. In muscle, a different phosphatase may have the role played by PP1 in liver, activating glycogen synthase by dephosphorylating it.
As we saw in Chapter 12, one way in which insulin triggers intracellular changes is by activating a protein kinase (PKB) that, in turn, phosphorylates and inactivates GSK3 (see Fig. 12-23). Phosphorylation of a Ser residue near the amino terminus of GSK3 converts that region of the protein to a pseudosubstrate, which folds into the site at which the priming phosphorylated Ser residue normally binds (Fig. 15-15b). This prevents GSK3 from binding the priming site of a real substrate, thereby inactivating the enzyme and tipping the balance in favor of dephosphorylation of glycogen synthase by PP1. Glycogen phosphorylase can also affect the phosphorylation of glycogen synthase: active glycogen phosphorylase directly inhibits PP1, preventing it from activating glycogen synthase (Fig. 15-14).
Insulin stimulates glycogen synthesis by activating PP1 and by inactivating GSK3. This single enzyme, PP1, can remove phosphoryl groups from all three of the enzymes phosphorylated in response to glucagon (liver) and epinephrine (liver and muscle): phosphorylase kinase, glycogen phosphorylase, and glycogen synthase. The catalytic subunit of PP1 (PP1c) does not exist free in the cytosol, but is tightly bound to its target proteins through a tissue-specific regulatory subunit, one of a family of glycogen-targeting proteins that bind glycogen and each of the three enzymes (Fig. 15-16). PP1 is itself subject to covalent and allosteric regulation: it is inactivated when phosphorylated by PKA and is allosterically activated by glucose 6-phosphate.
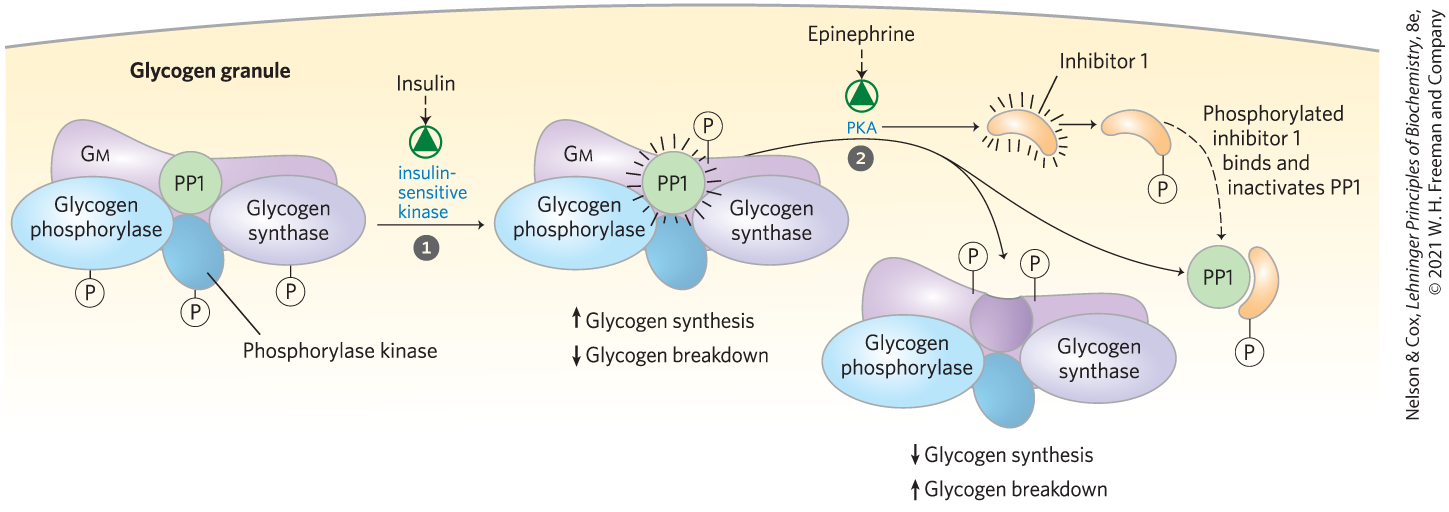
FIGURE 15-16 Glycogen-targeting protein Gm. Gm is a regulatory subunit of PP1 in muscle, and one of a family of glycogen-targeting proteins that serve as a scaffold, binding other proteins (including PP1) to glycogen particles. Gm can be phosphorylated at two different sites in response to insulin or epinephrine. Insulin-stimulated phosphorylation of Gm site 1 activates PP1, which dephosphorylates phosphorylase kinase, glycogen phosphorylase, and glycogen synthase. Epinephrine-stimulated phosphorylation of Gm site 2 by PKA causes dissociation of PP1 from the glycogen particle, preventing its access to glycogen phosphorylase and glycogen synthase. PKA also phosphorylates a protein (inhibitor 1) that, when phosphorylated, inhibits PP1. By these means, insulin stimulates glycogen synthesis and inhibits glycogen breakdown, whereas epinephrine (or glucagon in the liver) has the opposite effects.
 Insulin-stimulated phosphorylation of G
Insulin-stimulated phosphorylation of G Epinephrine-stimulated phosphorylation of G
Epinephrine-stimulated phosphorylation of GAllosteric and Hormonal Signals Coordinate Carbohydrate Metabolism Globally
Having looked at the mechanisms that regulate individual enzymes, we can now consider the overall shifts in carbohydrate metabolism that occur in the well-fed state, during fasting, and in the fight-or-flight response — signaled by insulin, glucagon, and epinephrine, respectively. We need to contrast two cases in which regulation serves different ends: (1) the role of hepatocytes in supplying glucose to the blood; and (2) the selfish use of carbohydrate fuels by extrahepatic tissues, typified by skeletal muscle (myocytes), to support their own activities.
After ingestion of a carbohydrate-rich meal, the elevation of blood glucose triggers insulin release (Fig. 15-17, top). In a hepatocyte, insulin has two immediate effects: it inactivates GSK3; and it activates a protein phosphatase, probably PP1. These two actions fully activate glycogen synthase. PP1 also inactivates glycogen phosphorylase a and phosphorylase kinase by dephosphorylating both, effectively stopping glycogen breakdown. Glucose enters the hepatocyte through the high-capacity transporter GLUT2, always present in the plasma membrane, and the elevated intracellular glucose leads to dissociation of hexokinase IV (glucokinase) from its nuclear regulatory protein (Fig. 14-21). Hexokinase IV enters the cytosol and phosphorylates glucose, stimulating glycolysis and supplying the precursor for glycogen synthesis. Under these conditions, hepatocytes use the excess glucose in the blood to synthesize glycogen, up to the limit of about 10% of the total weight of the liver.
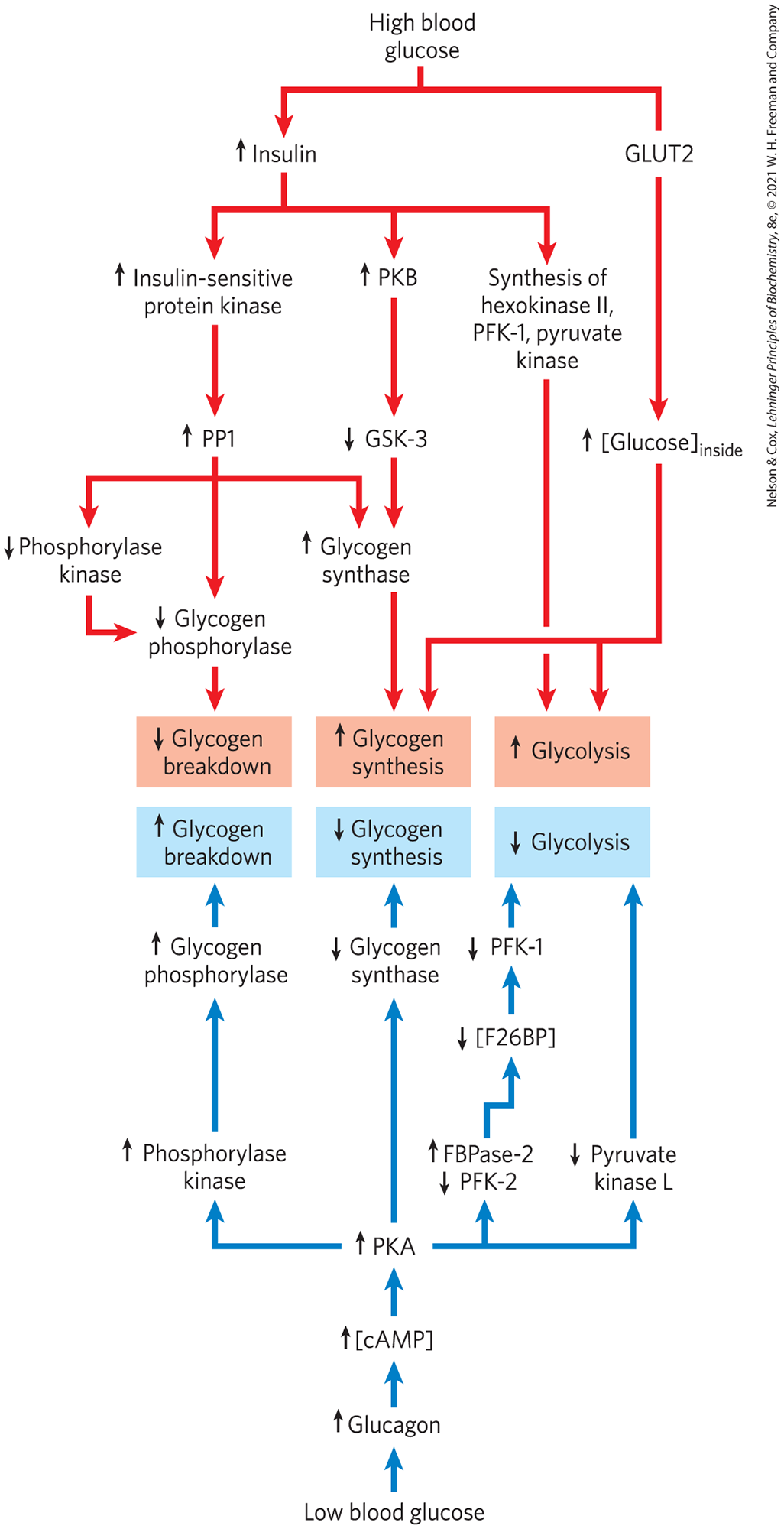
FIGURE 15-17 Regulation of carbohydrate metabolism in the liver. Colored arrows indicate causal relationships between the changes they connect. For example, an arrow from to means that a decrease in A causes an increase in B. Red arrows connect events that result from high blood glucose; blue arrows connect events that result from low blood glucose.
Between meals, or during an extended fast, the blood glucose level drops, triggering the release of glucagon, which, acting through the cascade shown in Figure 15-12, activates PKA. PKA mediates all the effects of glucagon (Fig. 15-17, bottom). It phosphorylates phosphorylase kinase, activating it and leading to the activation of glycogen phosphorylase. It phosphorylates glycogen synthase, inactivating it and blocking glycogen synthesis. It phosphorylates PFK-2/FBPase-2, leading to a drop in the concentration of the regulator fructose 2,6-bisphosphate, which has the effect of inactivating the glycolytic enzyme PFK-1 and activating the gluconeogenic enzyme FBPase-1 (see Fig. 14-24). And it phosphorylates and inactivates the glycolytic enzyme pyruvate kinase. Under these conditions, the liver produces glucose 6-phosphate by glycogen breakdown and by gluconeogenesis, and it stops using glucose to fuel glycolysis or make glycogen, maximizing the amount of glucose it can release to the blood. This release of glucose is possible only in liver and kidney, because other tissues lack glucose 6-phosphatase (Fig. 15-6).
The physiology of skeletal muscle differs from that of liver in three ways important to our discussion of metabolic regulation (Fig. 15-18): (1) muscle uses its stored glycogen only for its own needs; (2) as it goes from rest to vigorous contraction, muscle undergoes very large changes in its demand for ATP, which is provided by glycolysis; (3) muscle lacks the enzymatic machinery for gluconeogenesis. The regulation of carbohydrate metabolism in muscle reflects these differences from liver. First, myocytes lack receptors for glucagon, which are present on hepatocytes. Second, the muscle isozyme of pyruvate kinase is not phosphorylated by PKA, so glycolysis is not turned off when [cAMP] is high. In fact, cAMP increases the rate of glycolysis in muscle, probably by activating glycogen phosphorylase. When epinephrine is released into the blood in a fight-or-flight situation, PKA is activated by the rise in [cAMP] and phosphorylates and activates glycogen phosphorylase kinase. The resulting phosphorylation and activation of glycogen phosphorylase results in faster glycogen breakdown. Epinephrine is not released under low-stress conditions, but with each neuronal stimulation of muscle contraction, cytosolic rises briefly and activates phosphorylase kinase through its calmodulin subunit.
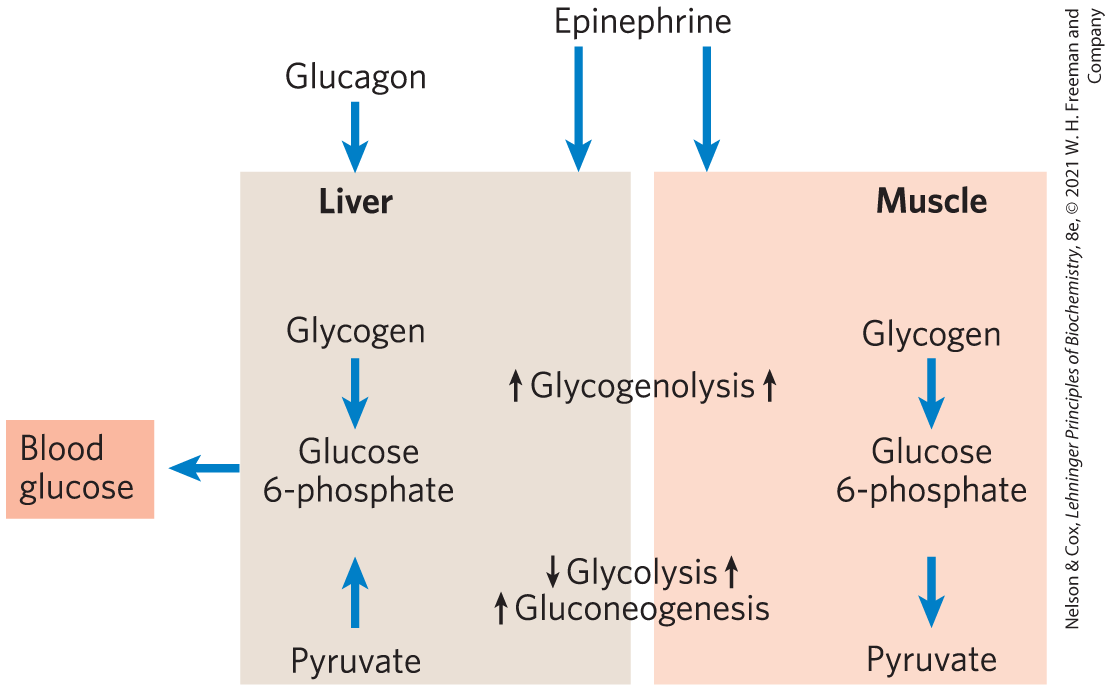
FIGURE 15-18 Difference in the regulation of carbohydrate metabolism in liver and muscle. In liver, either glucagon (indicating low blood glucose) or epinephrine (signaling the need to fight or flee) has the effect of maximizing the output of glucose into the blood. In muscle, epinephrine increases glycogen breakdown and glycolysis, which together provide fuel to produce the ATP needed for muscle contraction.
Elevated insulin triggers increased glycogen synthesis in myocytes by activating PP1 and inactivating GSK3. Unlike hepatocytes, myocytes have a reserve of the glucose transporter GLUT4 sequestered in intracellular vesicles. Insulin triggers their movement to the plasma membrane (see Fig. 12-23), where they allow increased glucose uptake. In response to insulin, therefore, myocytes help to lower blood glucose by increasing their rates of glucose uptake, glycogen synthesis, and glycolysis.
Carbohydrate and Lipid Metabolism Are Integrated by Hormonal and Allosteric Mechanisms
As complex as the regulation of carbohydrate metabolism is, it is far from the whole story of fuel metabolism. The metabolism of fats and fatty acids is very closely tied to that of carbohydrates. Hormonal signals such as insulin and changes in diet or exercise are equally important in regulating fat metabolism and integrating it with that of carbohydrates. We return to this overall metabolic integration in mammals in Chapter 23, after first considering the metabolic pathways for fats and amino acids (Chapters 17 and 18). The message we wish to convey here is that metabolic pathways are overlaid with complex regulatory controls that are exquisitely sensitive to changes in metabolic circumstances. These mechanisms act to adjust the flow of metabolites through various metabolic pathways, as needed by the cell and organism, without causing major changes in the concentrations of intermediates shared with other pathways.
SUMMARY 15.3 Coordinated Regulation of Glycogen Breakdown and Synthesis
- Glycogen phosphorylase is activated in response to epinephrine (in muscle) or glucagon (in liver), which raise [cAMP] and thereby activate PKA. PKA phosphorylates and activates phosphorylase kinase, which converts glycogen phosphorylase b to its active a form. In muscle, and AMP act allosterically to amplify the activity of the enzymes in this cascade. In liver, glucose acts allosterically to make phosphorylase a more susceptible to dephosphorylation/inactivation by phosphoprotein phosphatase 1 (PP1).
- Glycogen synthase a is inactivated by phosphorylation, ultimately catalyzed by GSK3, but primed by phosphorylation by other kinases. Insulin, by causing inactivation of GSK3 and activating PP1, favors glycogen synthase a and stimulates glycogen synthesis. Glucose 6-phosphate acts allosterically to make glycogen synthase b a better substrate for PP1, which dephosphorylates it to its active a form.
- In liver, glucagon stimulates glycogen breakdown and gluconeogenesis while blocking glycolysis, thereby sparing glucose for export to the brain and other tissues. In muscle, epinephrine stimulates glycogen breakdown and glycolysis, providing ATP to support contraction.
- Further hormonal and allosteric mechanisms regulate the use of carbohydrates and lipids as metabolic fuels.
 Glycogen phosphorylase is activated in response to epinephrine (in muscle) or glucagon (in liver), which raise [cAMP] and thereby activate PKA. PKA phosphorylates and activates phosphorylase kinase, which converts glycogen phosphorylase b to its active a form. In muscle, and AMP act allosterically to amplify the activity of the enzymes in this cascade. In liver, glucose acts allosterically to make phosphorylase a more susceptible to dephosphorylation/inactivation by phosphoprotein phosphatase 1 (PP1).
Glycogen phosphorylase is activated in response to epinephrine (in muscle) or glucagon (in liver), which raise [cAMP] and thereby activate PKA. PKA phosphorylates and activates phosphorylase kinase, which converts glycogen phosphorylase b to its active a form. In muscle, and AMP act allosterically to amplify the activity of the enzymes in this cascade. In liver, glucose acts allosterically to make phosphorylase a more susceptible to dephosphorylation/inactivation by phosphoprotein phosphatase 1 (PP1).