12.2 G Protein–Coupled Receptors and Second Messengers
As their name implies, G protein–coupled receptors (GPCRs) are receptors that act through a member of the guanosine nucleotide–binding protein, or G protein, family. Three essential components define signal transduction through GPCRs: a plasma membrane receptor with seven transmembrane helical segments, a G protein that cycles between active (guanosine triphosphate (GTP)-bound) and inactive (guanosine diphosphate (GDP)-bound) forms, and an effector enzyme (or ion channel) in the plasma membrane that is regulated by the activated G protein. An extracellular signal such as a hormone, growth factor, or neurotransmitter is the “first messenger” that activates a receptor from outside the cell. Ligand binding to the receptor forces an allosteric transition that allows the receptor to interact with a G protein, causing it to exchange its bound GDP for a GTP from the cytosol. The G protein then dissociates from the activated receptor and binds to the nearby effector enzyme, altering its activity. The effector enzyme then causes a change in the cytosolic concentration of a low molecular weight metabolite (such as ,-cyclic AMP) or inorganic ion , which acts as a second messenger to activate or inhibit one or more downstream targets, often protein kinases.
The human genome encodes just over 800 GPCRs, about 350 for detecting hormones, growth factors, and other endogenous ligands, and up to 500 that serve as olfactory (smell) and gustatory (taste) receptors. The largest superfamily of proteins encoded in the human genome, GPCRs have been implicated in many common medical conditions, including allergies, depression, blindness, diabetes, and various cardiovascular defects. GPCR mutations are also found in 20% of all cancers. In the United States, more than a third of all pharmaceuticals on the market target a GPCR. For example, the β-adrenergic receptor, which mediates the effects of epinephrine, is the target of the “beta blockers,” prescribed for such diverse conditions as hypertension, cardiac arrhythmia, glaucoma, anxiety, and migraine headache. More than 100 of the GPCRs found in the human genome are still “orphan receptors,” meaning that their natural ligands are not yet identified, and so we know little about their biology. The β-adrenergic receptor, with well-understood biology and pharmacology, is the prototype for all GPCRs, and our discussion of signal-transducing systems begins there.
The β-Adrenergic Receptor System Acts through the Second Messenger cAMP
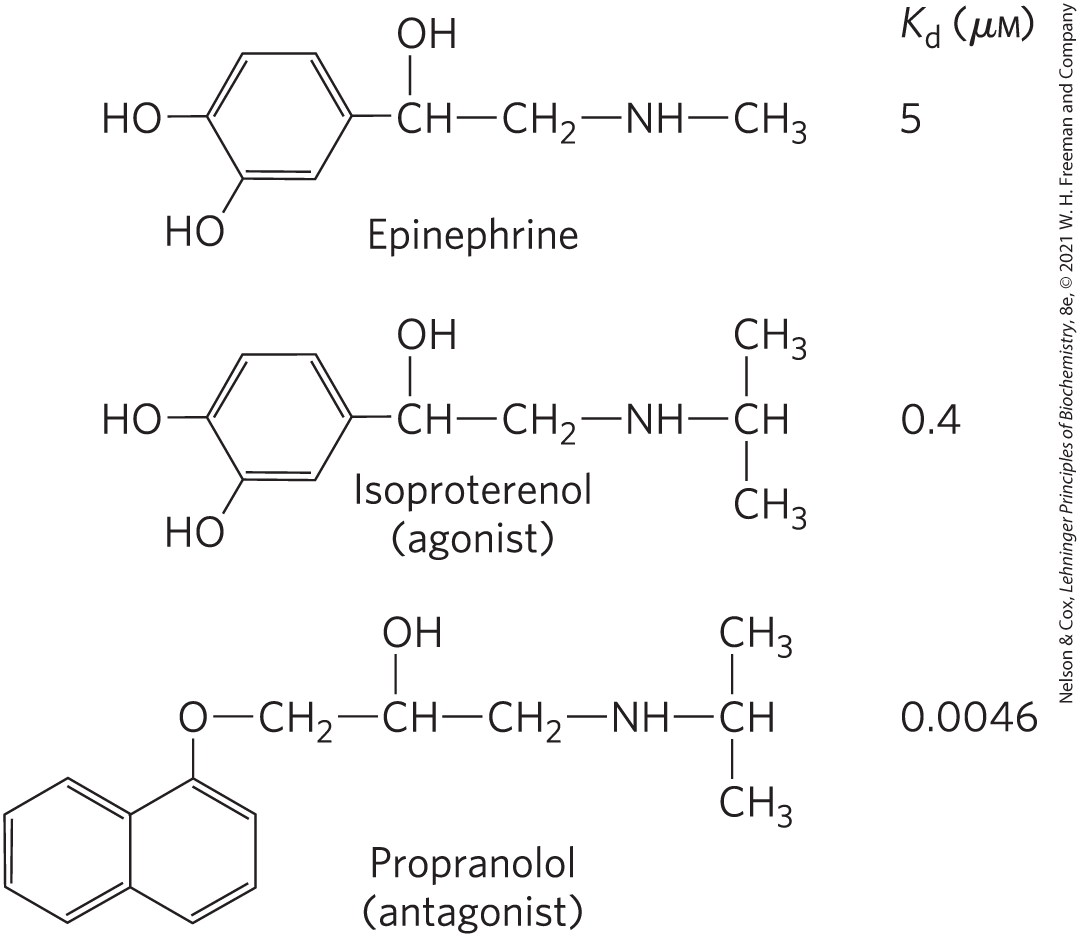
Epinephrine released from the adrenal glands sounds the alarm when a threat requires an animal to mobilize its energy-generating machinery; it signals the need to fight or flee. Epinephrine action begins when the hormone binds to a protein receptor in the plasma membrane of an epinephrine-sensitive cell (a myocyte in muscle, for example). Adrenergic receptors (“adrenergic” reflects the alternative name for epinephrine, adrenaline) are of four general types, , , , and , defined by differences in their affinities and responses to a group of agonists and antagonists. Agonists are molecules (natural ligands or their structural analogs) that bind a receptor and produce the effects of the natural ligand; antagonists are analogs that bind the receptor without triggering the normal effect and thereby block the effects of agonists, including the natural ligand. In some cases, the affinity of a synthetic agonist or antagonist for the receptor is greater than that of the natural agonist (Fig. 12-3). The four types of adrenergic receptors are found in different target tissues and mediate different responses to epinephrine. Here we focus on the β-adrenergic receptors of muscle, liver, and adipose tissue. These receptors mediate changes in fuel metabolism, as described in Chapter 23, including the increased breakdown of glycogen and fat. Adrenergic receptors of the and subtypes act through the same mechanism, so in our discussion, “β-adrenergic” applies to both types.
FIGURE 12-3 Epinephrine and its synthetic analogs. Epinephrine regulates energy-yielding metabolism in muscle, liver, and adipose tissue. Its affinity for its receptor is expressed as a dissociation constant for the receptor-ligand complex. Isoproterenol and propranolol are synthetic analogs, one an agonist with an affinity for the receptor that is higher than that of epinephrine, and the other an antagonist with extremely high affinity.
Like all GPCRs, the β-adrenergic receptor is an integral protein with seven hydrophobic, helical regions of 20 to 28 amino acid residues that span the plasma membrane seven times, thus the alternative names for GPCRs: seven-transmembrane (7tm) or heptahelical receptors. GPCRs effect signal transduction through interaction with heterotrimeric G proteins, a conserved family of signaling proteins with three subunits, α, β, and γ. The binding site for GDP or GTP is on the α subunit. When GDP is bound, the G protein is in its trimeric, inactive form.
GPCRs are allosteric proteins. When epinephrine binds to the β-adrenergic receptor (Fig. 12-4a, step ), allosteric transitions in the receptor and its associated G protein favor the replacement of GDP with GTP. Thus the hormone-bound GPCR acts as a guanosine nucleotide–exchange factor (GEF). In this active form, the G protein (step ) can transmit the signal from the activated receptor to the downstream effector protein, adenylyl cyclase. Because this G protein stimulates its effector, it is referred to as a stimulatory G protein, or . In the active form, the β and γ subunits of dissociate from the α subunit as a βγ dimer, and , with its bound GTP, moves in the plane of the membrane from the receptor to a nearby molecule of adenylyl cyclase (step ).
FIGURE 12-4 Transduction of the epinephrine signal: the β-adrenergic pathway. (a) The mechanism that couples binding of epinephrine to its receptor with activation of adenylyl cyclase; the seven steps are discussed in the text. The same adenylyl cyclase molecule in the plasma membrane may be regulated by a stimulatory G protein , as shown, or by an inhibitory G protein (, not shown). and are under the influence of different hormones. Hormones that induce GTP binding to cause inhibition of adenylyl cyclase, resulting in lower cellular [cAMP]. (b) The combined action of the enzymes that catalyze steps and , synthesis and hydrolysis of cAMP by adenylyl cyclase and cAMP phosphodiesterase, respectively.
Adenylyl cyclase is an integral protein of the plasma membrane, with its active site on the cytoplasmic face. The association of active with adenylyl cyclase stimulates the cyclase to catalyze the synthesis of second messenger cAMP from ATP (Fig. 12-4a, step ; Fig. 12-4b), raising the cytosolic [cAMP]. The interaction between and adenylyl cyclase occurs only when is bound to GTP.
The stimulation by is self-limiting; has intrinsic GTPase activity that switches to its inactive form by converting its bound GTP to GDP (Fig. 12-5). The inactive dissociates from adenylyl cyclase, rendering the cyclase inactive. reassociates with the βγ dimer , and inactive is again available to interact with a hormone-bound receptor.
FIGURE 12-5 The GTPase switch. G proteins cycle between GDP-bound (off) and GTP-bound (on). The protein’s GTPase activity, in many cases stimulated by RGS proteins (regulators of G-protein signaling; see Fig. 12-8), determines how quickly bound GTP is hydrolyzed to GDP and thus how long the G protein remains active.
Cyclic AMP Activates Protein Kinase A
Epinephrine exerts its downstream effects through the increase in [cAMP] that results from activation of adenylyl cyclase. Cyclic AMP, the second messenger, allosterically activates cAMP-dependent protein kinase, also called protein kinase A or PKA (Fig. 12-4a, step ), which catalyzes the phosphorylation of specific Ser or Thr residues of targeted proteins, such as glycogen phosphorylase b kinase. The latter enzyme is active when phosphorylated and can begin the process of mobilizing glycogen stores in muscle and liver in anticipation of the need for energy, as signaled by epinephrine.
The inactive form of PKA has two identical catalytic subunits (C) and two identical regulatory subunits (R) (Fig. 12-6a). The tetrameric complex is catalytically inactive, because an autoinhibitory domain of each R subunit occupies the substrate-binding cleft of each C subunit. The autoinhibitory domain is an intrinsically disordered region that can fold to fit any of several protein partners. Cyclic AMP is an allosteric activator of PKA. When cAMP binds to the R subunits, they undergo a conformational change that moves the autoinhibitory domain of R out of the catalytic domain of C, and the complex dissociates to yield two free, catalytically active C subunits. This same basic mechanism — displacement of an autoinhibitory domain — mediates the allosteric activation of many types of protein kinases by their second messengers (as in Fig. 12-21 and 12-29, for example). The structure of the substrate-binding cleft in PKA is the prototype for all known protein kinases (Fig. 12-6b); certain residues in this cleft region have identical counterparts in all of the 544 protein kinases encoded in the human genome. The ATP-binding site of each catalytic subunit positions ATP perfectly for the transfer of its terminal phosphoryl group to the in the side chain of a Ser or Thr residue in the target protein.
FIGURE 12-6 Activation of cAMP-dependent protein kinase (PKA). (a) When [cAMP] is low, the two identical regulatory subunits (R; red) associate with the two identical catalytic subunits (C; blue). In this complex, the inhibitor sequences of the R subunits lie in the substrate-binding cleft of the C subunits and prevent binding of protein substrates; the complex is therefore catalytically inactive. The amino-terminal sequences of the R subunits interact to form an dimer, the site of binding to an A kinase anchoring protein (AKAP; Fig. 12-11). When [cAMP] rises in response to a hormonal signal, each R subunit binds two cAMP molecules and undergoes a dramatic reorganization that pulls its inhibitory sequence away from the C subunit, opening up the substrate-binding cleft and releasing each C subunit in its catalytically active form. (b) A crystal structure showing part of the complex — one C subunit and part of one R subunit. The amino-terminal dimerization region of the R subunit is omitted for simplicity. The small lobe of C contains the ATP-binding site, and the large lobe surrounds and defines the cleft where the protein substrate binds and undergoes phosphorylation at a Ser or Thr residue, with a phosphoryl group transferred from ATP. In this inactive form, the inhibitor sequence of R blocks the substrate-binding cleft of C, inactivating it. [(b) Data from PDB ID 3FHI, C. Kim et al., Science 307:690, 2005.]
As indicated in Figure 12-4a (step ), PKA regulates many enzymes downstream in the signaling pathway. Although these downstream targets have diverse functions, they share a region of sequence similarity around the Ser or Thr residue that undergoes phosphorylation, a sequence that marks them for regulation by PKA. The substrate-binding cleft of PKA recognizes these sequences and phosphorylates their Thr or Ser residue. Comparison of the sequences of various protein substrates for PKA has yielded the short consensus sequence in which the target for phosphorylation (Ser or Thr) is most commonly embedded. For PKA, the consensus sequence is xR[RK]x[ST]B, where x can be any residue, R is Arg, [RK] can be either Arg or Lys, [ST] is either Ser or Thr (which is the residue phosphorylated), and B is any basic residue. The consensus sequences of a number of protein kinases are shown in Table 6-10; they define the targets of the hundreds of protein kinases in the eukaryotic cell.
How do we know that two proteins interact, or where in the cell they interact? Changes in the state of association of two proteins (such as the R and C subunits of PKA) can be seen by measuring the nonradiative transfer of energy between fluorescent probes attached to each protein, a technique called fluorescence resonance energy transfer (FRET), which we describe in Box 12-1.
As in many signaling pathways, signal transduction by the β-adrenergic receptor and adenylyl cyclase entails several steps that amplify the original hormone signal (Fig. 12-7). First, the binding of one hormone molecule to one receptor molecule catalytically activates many molecules that associate with the activated receptor, one after the other. Next, by activating one molecule of adenylyl cyclase, each active molecule stimulates the catalytic synthesis of many molecules of cAMP. The second messenger cAMP now activates PKA, each molecule of which catalyzes the phosphorylation of many molecules of the target protein — phosphorylase b kinase in Figure 12-7. This kinase activates glycogen phosphorylase b, which leads to the rapid mobilization of glucose from glycogen. The net effect of the cascade is amplification of the hormonal signal by orders of magnitude, which accounts for the very low concentration of epinephrine (or any other hormone) required for hormone activity. This signaling pathway is also rapid: the signal leads to intracellular changes within fractions of a second.
FIGURE 12-7 Epinephrine cascade. Epinephrine triggers a series of reactions in hepatocytes in which catalysts activate catalysts, resulting in great amplification of the original hormone signal. The numbers of molecules shown are simply to illustrate amplification and are almost certainly gross underestimates. Binding of one molecule of epinephrine to one β-adrenergic receptor on the cell surface activates many (possibly hundreds of) G proteins, one after another, each of which goes on to activate a molecule of the enzyme adenylyl cyclase. Adenylyl cyclase acts catalytically, producing many molecules of cAMP for each activated adenylyl cyclase. (Because two molecules of cAMP are required to activate one PKA catalytic subunit, this step does not amplify the signal.)
Several Mechanisms Cause Termination of the β-Adrenergic Response
To be useful, a signal-transducing system has to turn off after the hormonal or other stimulus has ended, and mechanisms for shutting off the signal are part of all signaling systems. Most systems also adapt to the continued presence of the signal by becoming less sensitive to it, by desensitizing. The β-adrenergic system illustrates both. Here, our focus is on termination.
The response to β-adrenergic stimulation will end when the concentration of the ligand (epinephrine) in the blood drops below the for its receptor. The epinephrine then dissociates from the receptor, and the latter reassumes its inactive conformation, in which it can no longer activate .
A second means of ending the response is the hydrolysis of GTP bound to the subunit, catalyzed by the GTPase activity of the G protein. Conversion of bound GTP to GDP favors the return of to the conformation in which it binds the subunits — the conformation in which the G protein is unable to interact with or stimulate adenylyl cyclase. This ends the production of cAMP. The rate of inactivation of depends on the GTPase activity, which for alone is very feeble. However, GTPase activator proteins (GAPs) strongly stimulate this GTPase activity, causing more-rapid inactivation of the G protein (Fig. 12-8). GAPs can themselves be regulated by other factors, providing a fine-tuning of the response to β-adrenergic stimulation. A third mechanism for terminating the response is to remove the second messenger: cAMP is hydrolyzed to -AMP (which is not active as a second messenger) by cyclic nucleotide phosphodiesterase (Fig. 12-4a, step ; 12-4b).
FIGURE 12-8 Factors that regulate G-protein activity. Inactive G proteins, both small G proteins such as Ras and heterotrimeric G proteins such as , interact with upstream GTP-GDP exchange factors (red). Often these exchange factors are activated (*) receptors such as rhodopsin (Rh) and β-adrenergic receptors (AR). The G proteins are activated by GTP binding, and in the GTP-bound form, activate downstream effector enzymes (blue), such as cGMP phosphodiesterase (PDE), adenylyl cyclase (AC), and Raf. By modulating the GTPase activity of G proteins, GTPase activator proteins (GAPs, in the case of small G proteins) and regulators of G-protein signaling (RGSs) (yellow), determine how long the G protein will remain active.
Finally, at the end of the signaling pathway, the metabolic effects that result from phosphorylation of target enzymes by PKA are reversed by the action of phosphoprotein phosphatases, which hydrolyze phosphorylated Tyr, Ser, or Thr residues, releasing inorganic phosphate . About 190 genes in the human genome encode phosphoprotein phosphatases, fewer than the number (about 540) that encode protein kinases, reflecting the relative promiscuity of the phosphoprotein phosphatases. A single phosphoprotein phosphatase (PP1) dephosphorylates some 200 different phosphoprotein targets, including the β-adrenergic receptor and other GPCRs. Some phosphatases are known to be regulated; others may be constantly active. When [cAMP] drops and PKA returns to its inactive form (step in Fig. 12-4a), the balance between phosphorylation and dephosphorylation is tipped toward dephosphorylation by these phosphatases.
The β-Adrenergic Receptor Is Desensitized by Phosphorylation and by Association with Arrestin
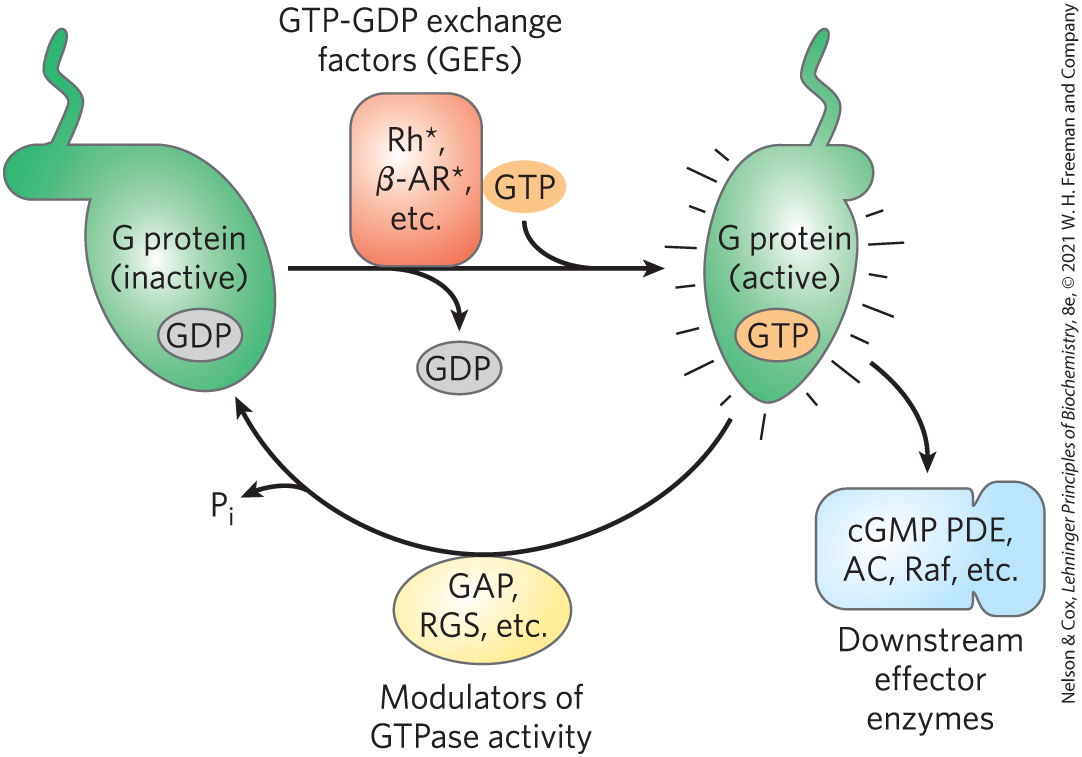
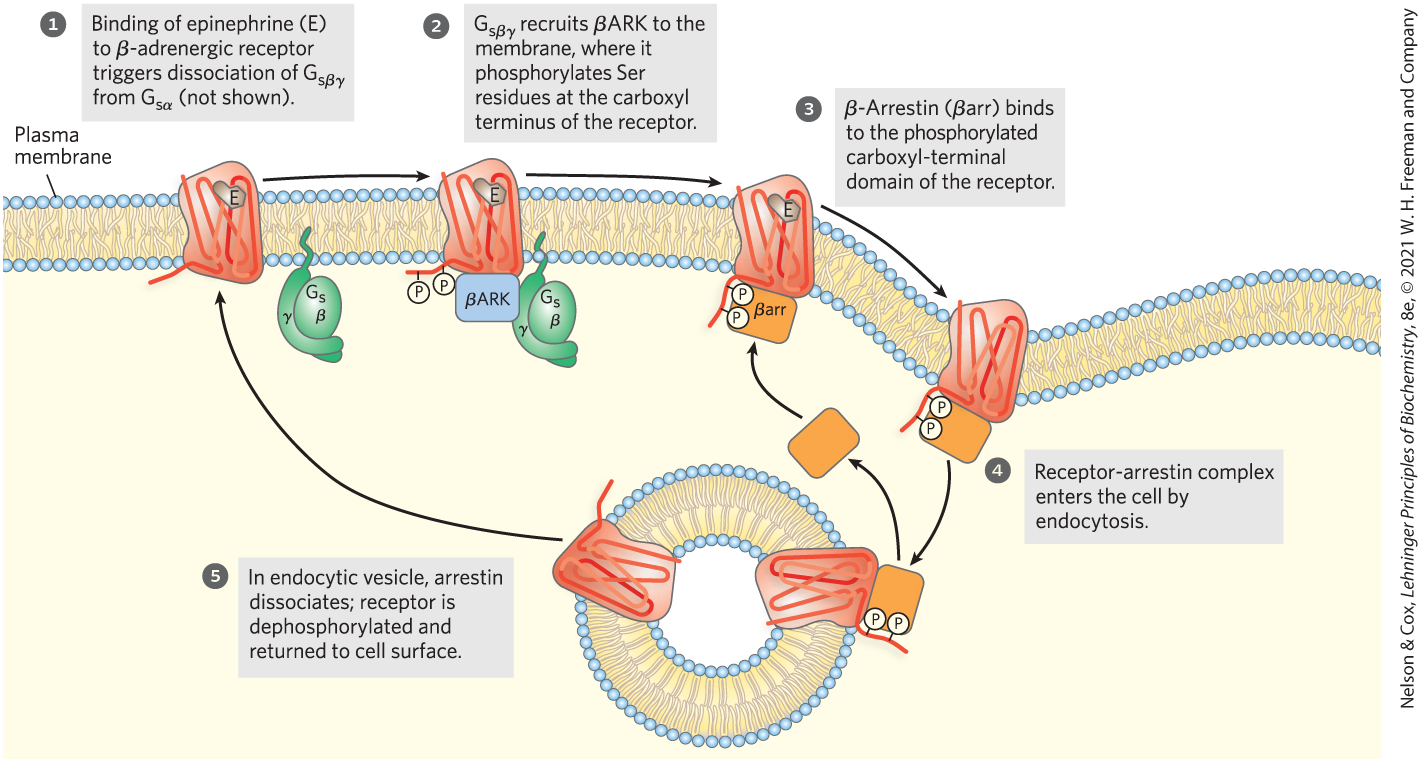
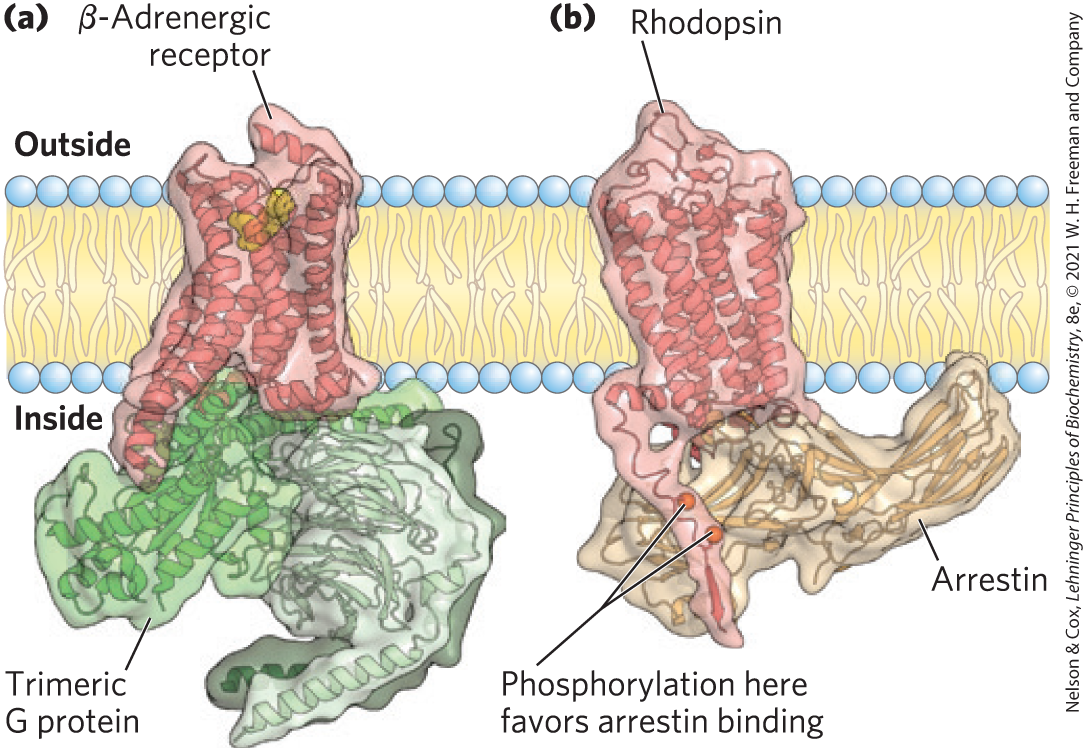
The mechanisms for signal termination described above take effect when the stimulus ends. A different mechanism, desensitization, damps the response even while the signal persists. When the receptor remains occupied with epinephrine, β-adrenergic receptor kinase, or βARK, phosphorylates several Ser residues near the receptor’s carboxyl terminus, which is on the cytoplasmic side of the plasma membrane (Fig. 12-9). βARK is drawn to the plasma membrane by its association with the subunits and is thus positioned to phosphorylate the receptor. Receptor phosphorylation creates a binding site for the protein β-arrestin, or βarr (also called arrestin 2), and binding of β-arrestin blocks the sites in the receptor that interact with the G protein (Fig. 12-10). The binding of β-arrestin also facilitates the sequestration of receptor molecules and their removal from the plasma membrane by endocytosis into small intracellular vesicles (endosomes). The arrestin-receptor complex recruits clathrin and other proteins involved in vesicle formation, which initiate membrane invagination, leading to the formation of endosomes containing the adrenergic receptor. In this state, the receptors are inaccessible to epinephrine and therefore inactive. These receptor molecules are eventually dephosphorylated and returned to the plasma membrane, completing the circuit and resensitizing the system to epinephrine.
FIGURE 12-9 Desensitization of the β-adrenergic receptor in the continued presence of epinephrine. This process is mediated by two proteins: β-adrenergic protein kinase (βARK) and β-arrestin (βarr). Not shown here is the phosphorylation and activation of βARK by PKA. PKA is activated by the rise in [cAMP] in response to the initial signal (epinephrine).
FIGURE 12-10 Mutual exclusion of trimeric G protein and arrestin in their interaction with a GPCR. (a) The β-adrenergic receptor complexed with its trimeric G protein, . (b) Another GPCR, the rhodopsin receptor, phosphorylated near its carboxyl terminus, and bound to arrestin. Binding of arrestin blocks binding and further activation of the G protein and ends the response to the initial signal. [Data from (a) PDB ID 3SN6, S. G. F. Rasmussen et al., Nature 477:549, 2011; (b) PDB ID 4ZWJ, Y. Kang et al., Nature 523:561, 2015.]
β-Adrenergic receptor kinase is a member of a family of G protein–coupled receptor kinases (GRKs), all of which phosphorylate GPCRs on their carboxyl-terminal cytoplasmic domains and play roles similar to that of βARK in desensitization and resensitization of their receptors. Seven GRKs and four different arrestins are encoded in the human genome; each GRK is capable of desensitizing a particular subset of GPCRs, and each arrestin can interact with many different types of phosphorylated receptors.
The receptor-arrestin complex has another important role: it initiates signaling by a second pathway, the MAPK cascade described below. Thus, acting through a single GPCR, epinephrine triggers two divergent signaling pathways. The two pathways, one initiated by the receptor’s interaction with a G protein and the other initiated by its interaction with arrestin, can be differentially affected by the agonist; in some cases, one agonist favors the G-protein pathway and another favors the arrestin pathway. This bias is an important consideration in the development of a medication that acts through a GPCR. For example, the most addictive of the opioid drugs of abuse act more strongly through G-protein signaling than through arrestin. An ideal opioid pain medication would act through the branch of the pathway that has therapeutic effects and not through the pathway that leads to addiction.
Cyclic AMP Acts as a Second Messenger for Many Regulatory Molecules
Epinephrine is just one of many hormones, growth factors, and other regulatory molecules that act by changing the intracellular [cAMP] and thus the activity of PKA. Table 12-3 lists a few examples. Glucagon binds to its receptors in the plasma membrane of adipocytes, activating (via a protein) adenylyl cyclase. PKA, stimulated by the resulting rise in [cAMP], phosphorylates and activates two proteins critical to the mobilization of the fatty acids of stored fats (see Fig. 17-2). Similarly, the peptide hormone ACTH (adrenocorticotropic hormone, also called corticotropin), produced by the anterior pituitary, binds to specific receptors in the adrenal cortex, activating adenylyl cyclase and raising the intracellular [cAMP]. PKA then phosphorylates and activates several of the enzymes required for the synthesis of cortisol and other steroid hormones. In many cell types, the catalytic subunit of PKA can also move into the nucleus, where it phosphorylates the cAMP response element binding protein (CREB), which alters the expression of specific genes regulated by cAMP.
TABLE 12-3 Some Signals That Use cAMP as Second Messenger
Corticotropin (ACTH)
Corticotropin-releasing hormone (CRH)
Dopamine [, ]
Epinephrine (β-adrenergic)
Follicle-stimulating hormone (FSH)
Glucagon
Histamine
Luteinizing hormone (LH)
Melanocyte-stimulating hormone (MSH)
Odorants (many)
Parathyroid hormone
Prostaglandins ,
Serotonin
Somatostatin
Tastants (sweet, bitter)
Thyroid-stimulating hormone (TSH)
Note: Receptor subtypes in square brackets. Subtypes may have different transduction mechanisms. For example, serotonin is detected in some tissues by receptor subtypes and , which act through adenylyl cyclase and cAMP, and in other tissues by receptor subtype , acting through the phospholipase mechanism (see Table 12-4).
Some hormones act by inhibiting adenylyl cyclase, thus lowering [cAMP] and suppressing protein phosphorylation by PKA. For example, the binding of somatostatin to its receptor in the pancreas leads to activation of an inhibitory G protein, or , structurally homologous to , that inhibits adenylyl cyclase and lowers [cAMP]. In this way, somatostatin inhibits the secretion of several hormones, including glucagon. In adipose tissue, prostaglandin (; see Fig. 10-17) inhibits adenylyl cyclase, thus lowering [cAMP] and slowing the mobilization of lipid reserves triggered by epinephrine and glucagon. In certain other tissues, stimulates cAMP synthesis: its receptors are coupled to adenylyl cyclase through a stimulatory G protein, . In tissues with -adrenergic receptors, epinephrine lowers [cAMP]; in this case, the receptors are coupled to adenylyl cyclase through an inhibitory G protein, . In short, an extracellular signal such as epinephrine or can have different effects on different tissues or cell types, depending on three factors: the type of receptor in the tissue, the type of G protein with which the receptor is coupled, and the set of PKA target enzymes in the cell. By summing the influences that tend to increase and decrease [cAMP], a cell achieves the integration of signals that is a general feature of signal-transducing mechanisms (Fig. 12-1f).
Another factor that explains how so many types of signals can be mediated by a single second messenger (cAMP) is the confinement of the signaling process to a specific region of the cell by adaptor proteins — noncatalytic proteins that hold together other protein molecules that function in concert (further described below). AKAPs (A kinase anchoring proteins) have multiple, distinct protein-binding domains, often intrinsically disordered regions; they are multivalent adaptor proteins. One domain binds to the R subunits of PKA and another binds to a specific structure in the cell, confining the PKA to the vicinity of that structure. For example, specific AKAPs bind PKA to microtubules, actin filaments, ion channels, mitochondria, or the nucleus. Different types of cells have different complements of AKAPs, so cAMP might stimulate phosphorylation of mitochondrial proteins in one cell and stimulate phosphorylation of actin filaments in another. In some cases, an AKAP connects PKA with the enzyme that triggers PKA activation (adenylyl cyclase) or terminates PKA action (cAMP phosphodiesterase or phosphoprotein phosphatase) (Fig. 12-11). The very close proximity of these activating and inactivating enzymes presumably achieves a highly localized, and very brief, response.
FIGURE 12-11 Nucleation of supramolecular complexes by A kinase anchoring proteins (AKAPs). AKAP5 is one of a family of proteins that act as multivalent scaffolds, holding PKA catalytic subunits — through interaction of the AKAP with the PKA regulatory subunits — in proximity to a particular region or structure in the cell. AKAP5 is targeted to rafts in the cytoplasmic face of the plasma membrane by two covalently attached palmitoyl groups and a site that binds phosphatidylinositol 3,4,5-trisphosphate in the membrane. AKAP5 also has binding sites for the β-adrenergic receptor, adenylyl cyclase, PKA, and a phosphoprotein phosphatase (PP2A), bringing them all together in the plane of the membrane. When epinephrine binds to the β-adrenergic receptor, adenylyl cyclase produces cAMP, which reaches the nearby PKA quickly and with very little dilution. PKA phosphorylates its target protein, altering its activity, until the phosphoprotein phosphatase removes the phosphoryl group and returns the target protein to its prestimulus state. The AKAPs in this and other cases bring about a high local concentration of enzymes and second messengers, so that the signaling circuit remains highly localized and the duration of the signal is limited.
In a process analogous with the cAMP-PKA pathway, a cyclic derivative of GTP (,-cyclic GMP; cGMP) is generated in response to an extracellular signal. The cGMP activates a cGMP-dependent protein kinase (PKG), which phosphorylates specific protein substrates, changing their activities in response to the initial signal (Box 12-2).
G Proteins Act as Self-Limiting Switches in Many Processes
Proteins sensitive to the binding of either GTP or GDP play critical roles in many cellular processes, including sensory perception, signaling for cell division, growth and differentiation, intracellular movements of proteins and membrane vesicles, and protein synthesis. The human genome encodes nearly 200 of these proteins, which differ in size and subunit structure, intracellular location, and function. But all G proteins share a common feature: they can become activated by binding GTP and then, after a brief period, can inactivate themselves with their GTPase activity, thereby serving as molecular binary switches with built-in timers. This superfamily of proteins includes the trimeric G proteins involved in β-adrenergic signaling ( or ) and vision (transducin); small, monomeric G proteins such as that involved in insulin signaling (Ras; see below) and others that function in vesicle trafficking (ARF, RAC1, and Rab), transport into and out of the nucleus (Ran), and timing of the cell cycle (Rho); and several proteins involved in protein synthesis (initiation factor IF2 and elongation factors EF-Tu and EF-G; see Chapter 27). Among the trimeric G proteins, subunits have covalently linked lipids: the amino terminus is palmitoylated, and some are also myristoylated. and the monomeric Ras protein have an isoprenyl lipid at their carboxyl termini. The attached lipids keep them anchored to the plasma membrane, restricting their action to that two-dimensional plane.
All G proteins have the same core structure and use the same mechanism for switching between an inactive conformation, favored when GDP is bound, and an active conformation, favored when GTP is bound. We can use Ras (~20 kDa, a minimal signaling unit) as a prototype for all members of this superfamily (Fig. 12-12).
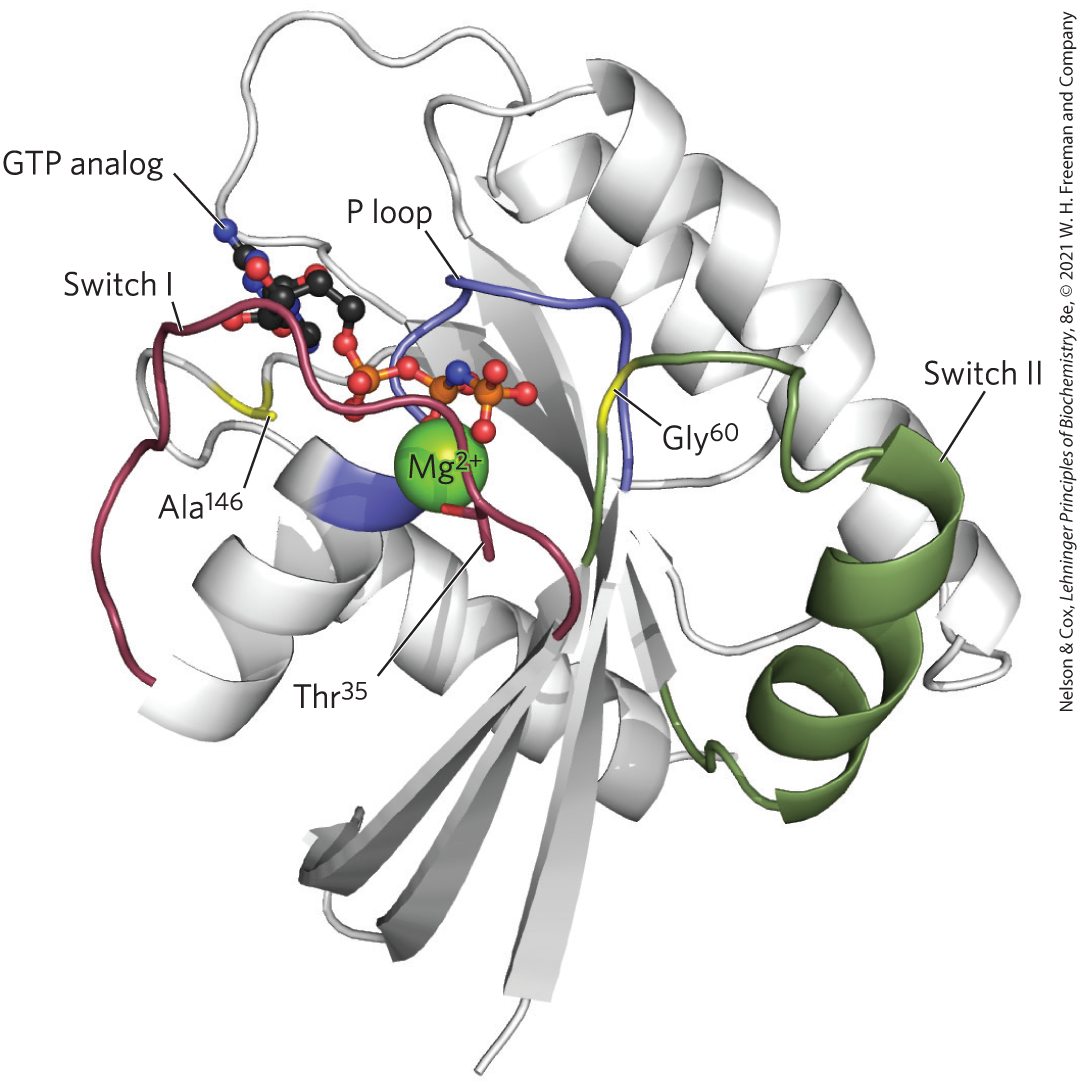
FIGURE 12-12 Ras, the G-protein prototype. -GTP is held by critical residues in the phosphate-binding P loop (blue) and by in the switch I region (red) and in the switch II region (green). gives specificity for GTP over ATP. In the structure shown here, the nonhydrolyzable GTP analog Gpp(NH)p is in the GTP-binding site. [Data from PDB ID 5P21, E. F. Pai et al., EMBO J. 9:2351, 1990.]
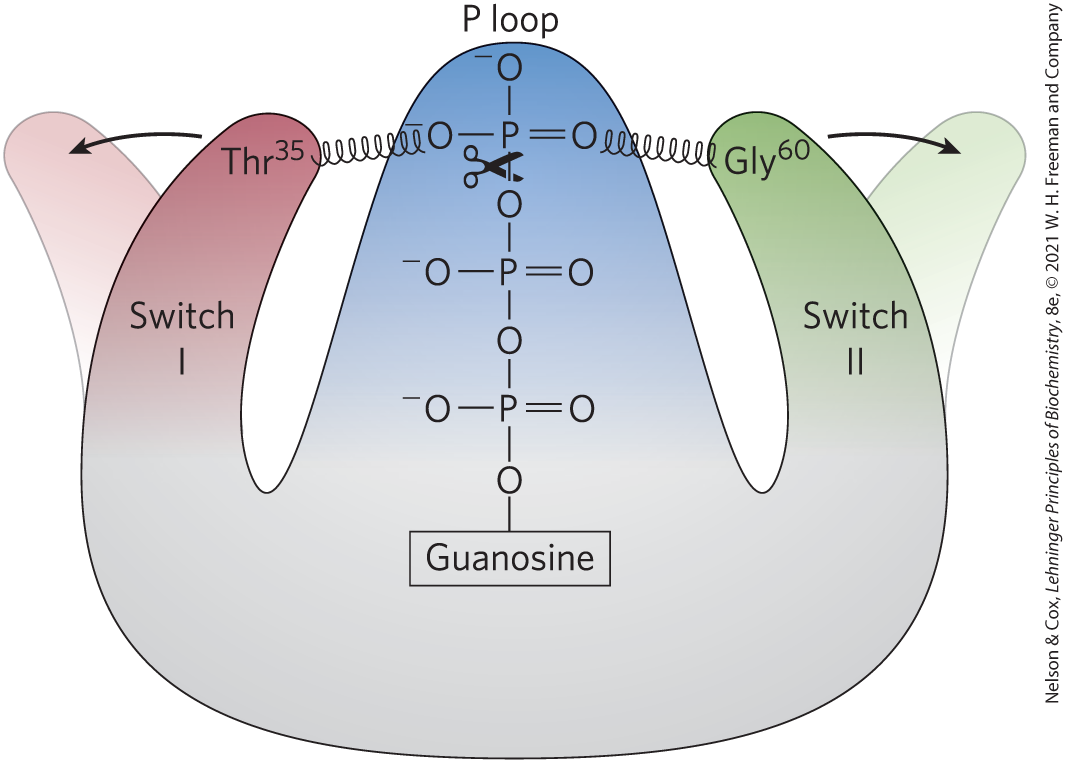
In the nucleotide-binding site of Ras, hydrogen bonds to the guanine oxygen, allowing GTP, but not ATP, to bind. In the GTP-bound conformation, the G protein exposes previously buried regions (called switch I and switch II) that interact with proteins downstream in the signaling pathway until the G protein inactivates itself by hydrolyzing its bound GTP to GDP. The critical determinant of G-protein conformation is the γ phosphate of GTP, which interacts with a region called the P loop (phosphate-binding). In Ras, the γ phosphate of GTP binds to a Lys residue in the P loop and to two critical residues, in switch I and in switch II, that hydrogen-bond with the oxygens of the γ phosphate of GTP. These hydrogen bonds act like a pair of springs holding the protein in its active conformation (Fig. 12-13). When GTP is cleaved to GDP, and is released, these hydrogen bonds are lost; the protein then relaxes into its inactive conformation, burying switch I and II so they are no longer available to interact with other partners.
FIGURE 12-13 GTP hydrolysis flips the switches in Ras. When bound GTP is hydrolyzed by the GTPase activities of Ras and its GTPase activator protein (GAP), loss of hydrogen bonds to and allows the switch I and switch II regions to relax into a conformation in which they are no longer available to interact with downstream targets. [Information from I. R. Vetter and A. Wittinghofer, Science 294:1299, 2001, Fig. 3.]
The GTPase activity of most G proteins is very weak, but it is increased up to -fold by GTPase activator proteins (GAPs), also called, in the case of heterotrimeric G proteins, regulators of G-protein signaling (RGSs; Fig. 12-8). GAPs and RGSs thus determine how long the G-protein switch remains on. There are about 40 different RGS proteins associated with a variety of processes and expressed in most tissues. They have a critical Arg residue that reaches into the G-protein GTPase active site and contributes to catalysis. The intrinsically slow process of replacing bound GDP with GTP, switching the protein on, is catalyzed by guanosine nucleotide–exchange factors (GEFs, such as the β-adrenergic receptor) associated with the G protein. The ligand-bound β-adrenergic receptor is one of many GEFs, and a broad range of proteins act as GAPs. Their combined effects set the level of GTP-bound G proteins, and thus the strength of the response to signals that arrive at the receptors.
Because G proteins play crucial roles in so many signaling processes, it is not surprising that defects in G proteins lead to a variety of diseases. In about 25% of all human cancers (and in a much higher proportion of certain types of cancer), a mutation in a Ras protein — typically in one of the critical residues around the GTP-binding site or in the P loop — virtually eliminates its GTPase activity. Once activated by GTP binding, this Ras protein remains constantly active, promoting cell division in cells that should not divide. The tumor suppressor gene NF1 encodes a GAP that enhances the GTPase activity of normal Ras. Mutations in NF1 that result in a nonfunctioning GAP leave Ras with only its GTPase activity, which is very weak (that is, has a very low turnover number); once activated by GTP binding, Ras stays active for an extended period, continuing to send the signal: divide.
Defective heterotrimeric G proteins can also lead to disease. Mutations in the gene that encodes the α subunit of (which mediates changes in [cAMP] in response to hormonal stimuli) may result in a that is permanently active or permanently inactive. “Activating” mutations generally occur in residues crucial to GTPase activity; they lead to a continuously elevated [cAMP], with significant downstream consequences, including undesirable cell proliferation. Such mutations are found in about 40% of pituitary tumors (adenomas). Individuals with “inactivating” mutations in are unresponsive to hormones (such as thyroid hormone) that act through cAMP. Mutation in the gene for the transducin α subunit , which is involved in visual signaling, leads to a type of night blindness, apparently due to defective interaction between the activated subunit and the phosphodiesterase of the rod outer segment (see Fig. 12-19). A sequence variation in the gene encoding the β subunit of a heterotrimeric G protein is commonly found in individuals with hypertension (high blood pressure), and this variant gene is suspected of involvement in obesity and atherosclerosis.
The pathogenic bacterium that causes cholera produces a toxin that targets a G protein, interfering with normal signaling in host cells. Cholera toxin, secreted by Vibrio cholerae in the intestine of an infected person, is a heterodimeric protein. Subunit B recognizes and binds to specific gangliosides on the surface of intestinal epithelial cells and provides a route for subunit A to enter these cells. After entry, subunit A is broken into two fragments, A1 and A2. A1 associates with the host cell’s ADP-ribosylation factor ARF6, a small G protein, through residues in its switch I and switch II regions — which are accessible only when ARF6 is in its active (GTP-bound) form. This association with ARF6 activates A1, which catalyzes the transfer of ADP-ribose from to the critical Arg residue in the P loop of the α subunit of (Fig. 12-14). ADP-ribosylation blocks the GTPase activity of and thereby renders permanently active. This results in continuous activation of the adenylyl cyclase of intestinal epithelial cells, chronically high [cAMP], and chronically active PKA. PKA phosphorylates the CFTR channel (see Box 11-2) and a exchanger in the intestinal epithelial cells. The resultant efflux of NaCl triggers massive water loss through the intestine as cells respond to the ensuing osmotic imbalance. Severe dehydration and electrolyte loss are the major pathologies in cholera. These can be fatal in the absence of prompt rehydration therapy.
FIGURE 12-14 ADP-ribosylation locks in the active conformation. The bacterial toxin that causes cholera is an enzyme that catalyzes transfer of the ADP-ribose moiety of (nicotinamide adenine dinucleotide) to an Arg residue of . The G proteins thus modified fail to respond to normal hormonal stimuli. The pathology of cholera results from defective regulation of adenylyl cyclase and overproduction of cAMP.
Diacylglycerol, Inositol Trisphosphate, and Have Related Roles as Second Messengers
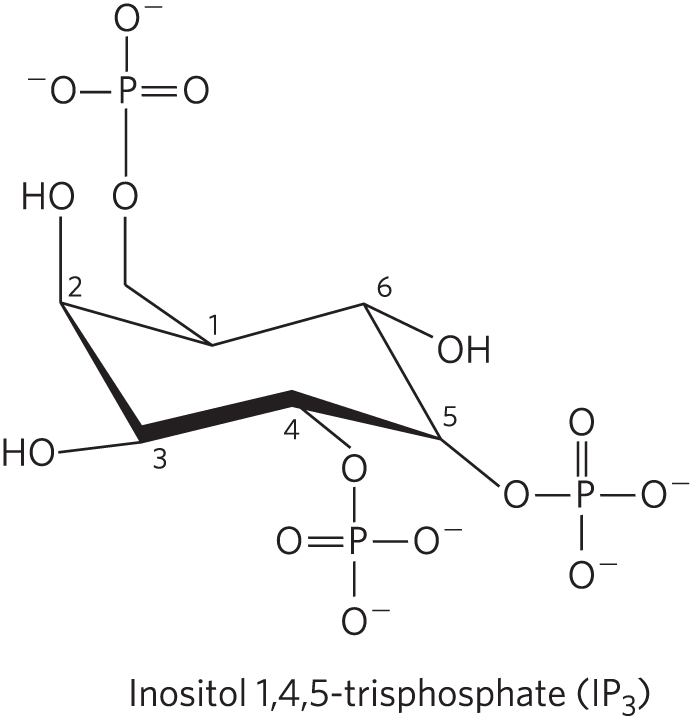
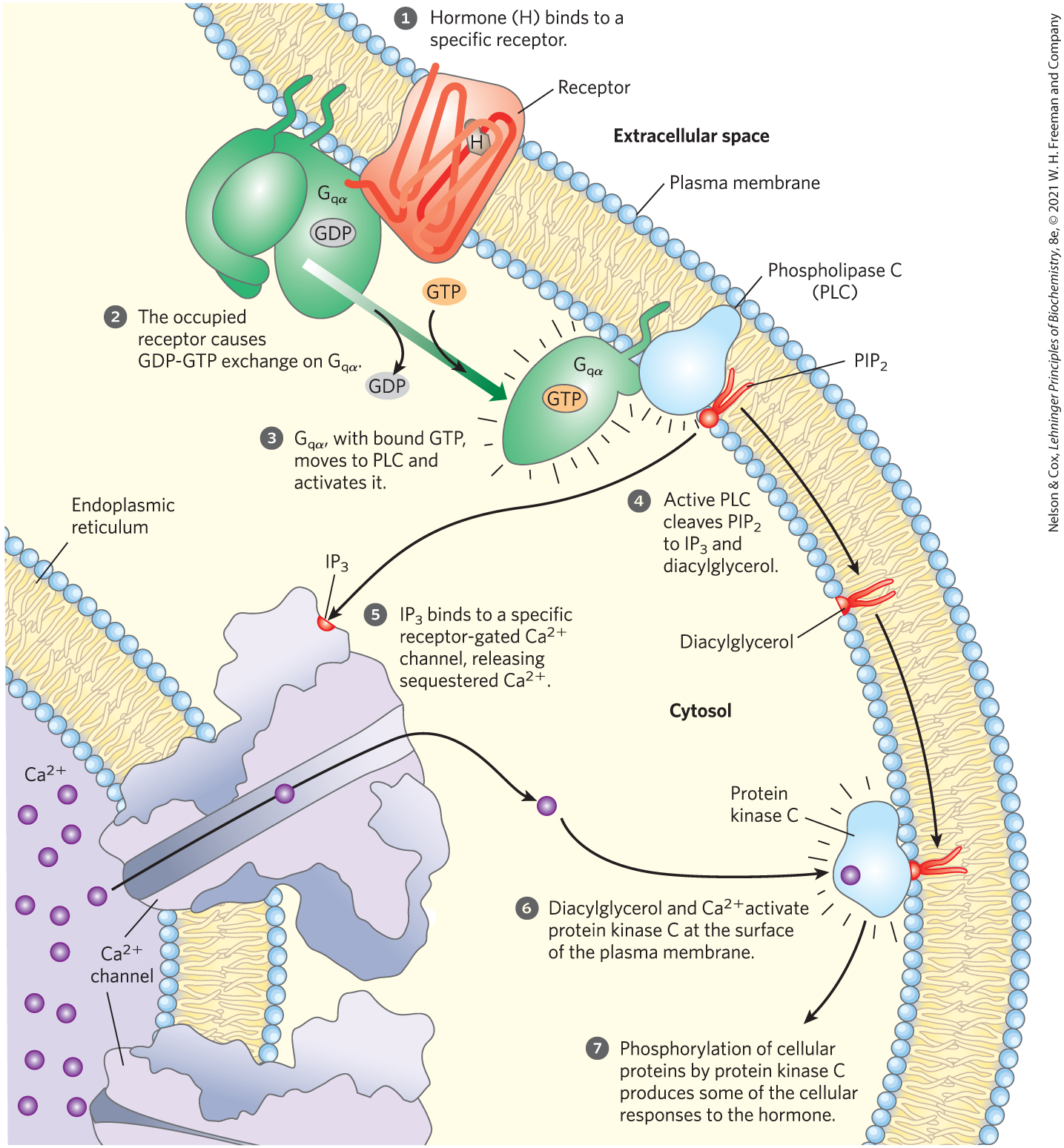
A second broad class of GPCRs is coupled through a G protein to a plasma membrane phospholipase C (PLC) that catalyzes cleavage of the membrane phospholipid phosphatidylinositol 4,5-bisphosphate, or (see Fig. 10-14). When one of the agonists (hormone, neurotransmitter, growth factor; Table 12-4) that act by this mechanism binds its specific receptor in the plasma membrane (Fig. 12-15, step ), the receptor-hormone complex catalyzes GTP-GDP exchange on an associated trimeric G protein, (step ). This activates the proteins in much the same way that the β-adrenergic receptor activates (Fig. 12-4). The activated activates the -specific PLC (Fig. 12-15, step ), which catalyzes the production of two potent second messengers (step ), diacylglycerol and inositol 1,4,5-trisphosphate, or (not to be confused with , p. 435 or Fig. 12-24).
TABLE 12-4 Some Signals That Act through Phospholipase C, , and
Acetylcholine [muscarinic ]
Gastrin-releasing peptide
Platelet-derived growth factor (PDGF)
-Adrenergic agonists
Glutamate
Serotonin
Angiogenin
Gonadotropin-releasing hormone (GRH)
Thyrotropin-releasing hormone (TRH)
Angiotensin II
Histamine
Vasopressin
ATP
Light (Drosophila)
Auxin
Oxytocin
Note: Receptor subtypes are in square brackets; see footnote to Table 12-3.
FIGURE 12-15 Hormone-activated phospholipase C and . Two intracellular second messengers are produced in the hormone-sensitive phosphatidylinositol system: inositol 1,4,5-trisphosphate and diacylglycerol are cleaved from phosphatidylinositol 4,5-bisphosphate . Both contribute to the activation of protein kinase C. By raising cytosolic , also activates other -dependent enzymes; thus also acts as a second messenger.
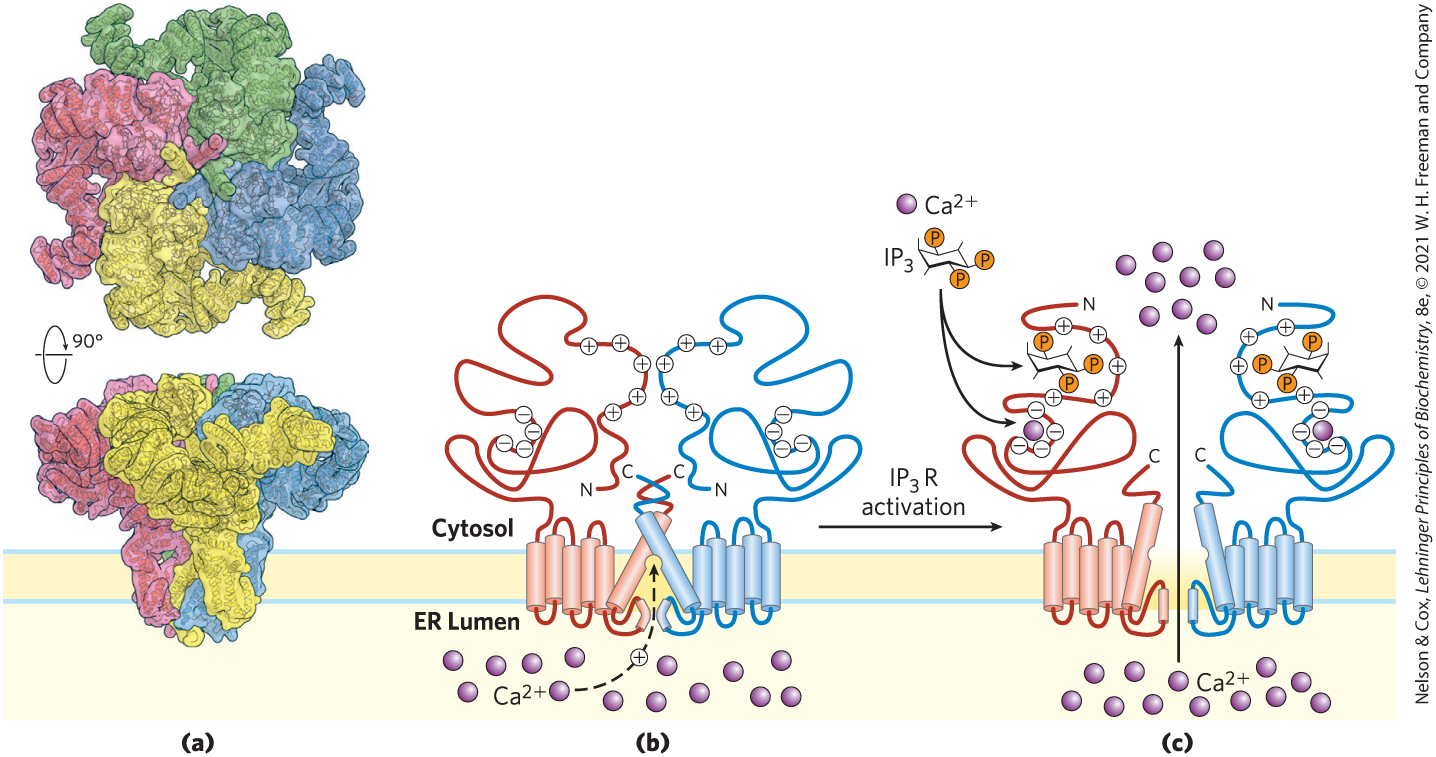
Inositol trisphosphate, a water-soluble compound, diffuses from the plasma membrane to the endoplasmic reticulum (ER), where it binds to the -gated channel, causing the channel to open. The action of the SERCA pump (pp. 393–394) ensures that in the ER is orders of magnitude higher than that in the cytosol, so when these gated channels open, rushes into the cytosol (Fig. 12-15, step ), and the cytosolic rises sharply to about . One effect of elevated is the activation of protein kinase C (PKC; C for ). Diacylglycerol cooperates with in activating PKC, thus also acting as a second messenger (step ). Activation involves the movement of a PKC domain (the pseudosubstrate domain) away from its location in the substrate-binding region of the enzyme, allowing the enzyme to bind and phosphorylate proteins that contain a PKC consensus sequence — Ser or Thr residues embedded in an amino acid sequence recognized by PKC (step ). Figure 12-16 shows the structure of the receptor and a proposed mechanism of its action as a gated channel.
FIGURE 12-16 Proposed mechanism of action of the -gated channel. (a) The receptor in its closed conformation, determined by cryo-EM. The 1.3 MDa tetramer has 24 transmembrane helices that surround a central pore. The bulk of the protein protrudes out of the ER into the cytosol and contains the -binding sites. The central pore is the channel for movement, which does not conduct in the absence of . (b, c) Model for receptor activation by , showing only two of the four identical subunits (b) in the absence of , and (c) with one bound to each subunit. According to this model, binding near the amino-terminal end of a subunit causes a major rearrangement of the α helix at the carboxyl-terminal end, opening the channel. [(a) Data from PDB ID 6MU2, G. Fan et al., Cell Res. 28:1158, 2018. (b, c) Information from M. J. Berridge, Physiol. Rev. 96:1261, 2016, Fig. 3.]
Several isozymes of PKC are known, each with a characteristic tissue distribution, target protein specificity, and role. PLCβ is activated by GPCRs; another isoform, PLCγ, is activated by receptor tyrosine kinases, as described below. The ultimate PKC targets include cytoskeletal proteins, enzymes, and nuclear proteins that regulate gene expression. Taken together, this family of enzymes has a wide range of cellular actions, affecting neuronal and immune function and the regulation of cell division. Compounds that lead to overexpression of PKC or that increase its activity to abnormal levels act as tumor promoters; animals exposed to these substances have increased rates of cancer.
Calcium Is a Second Messenger That Is Limited in Space and Time
There are many variations on this basic scheme for signaling. In many cell types that respond to extracellular signals, serves as a second messenger that triggers intracellular responses, such as exocytosis in neurons and endocrine cells, contraction in skeletal muscle, and cytoskeletal rearrangements during amoeboid movement. In unstimulated cells, cytosolic is kept very low by the action of pumps in the ER, mitochondria, and plasma membrane (as further discussed below). Hormonal, neural, or other stimuli cause either an influx of into the cell through specific channels in the plasma membrane or the release of sequestered from the ER or mitochondria, in either case raising the cytosolic and triggering a cellular response.
Changes in intracellular are detected by -binding proteins that regulate a variety of -dependent enzymes. Calmodulin (CaM; 17,000) is an acidic protein with four high-affinity -binding sites ( to 1 μm) (Fig. 12-17). When intracellular rises to about (1 μm), the binding of to calmodulin drives a conformational change in the protein. Calmodulin associates with a variety of proteins and, in its -bound state, modulates their activities. It is a member of a family of -binding proteins that also includes troponin (see Fig. 5-30), which triggers skeletal muscle contraction in response to increased . Members of this family share a characteristic -binding structure, the EF hand.
FIGURE 12-17 Calmodulin, the protein mediator of many -stimulated enzymatic reactions. (a) In this ribbon model of the crystal structure of calmodulin, the four high-affinity -binding sites are occupied by (purple). The amino-terminal domain is on the left; the carboxyl-terminal domain on the right. (b) Calmodulin associated with a helical domain of one of the many enzymes it regulates, calmodulin-dependent protein kinase II. Notice that the long central α helix of calmodulin visible in (a) has bent back on itself in binding to the helical substrate domain. The central helix of calmodulin is clearly more flexible in solution than in the crystal. (c) Each of the four -binding sites occurs in a helix-loop-helix motif called the EF hand, found in many -binding proteins. [Data from (a) PDB ID 1CLL, R. Chattopadhyaya et al., J. Mol. Biol. 228:1177, 1992; (b, c) PDB ID 1CDL, W. E. Meador et al., Science 257:1251, 1992.]
Calmodulin is a subunit of the /calmodulin-dependent protein kinases (CaM kinases, types I through IV). When intracellular increases in response to a stimulus, calmodulin binds , undergoes a change in conformation, and activates the CaM kinase. The kinase then phosphorylates target enzymes, regulating their activities. Calmodulin is also a regulatory subunit of phosphorylase b kinase of muscle, which is activated by . Thus triggers ATP-requiring muscle contractions while also activating glycogen breakdown, providing fuel for ATP synthesis. Many other enzymes are known to be modulated by through calmodulin (Table 12-5). The activity of the second messenger , like that of cAMP, can be spatially restricted; after its release triggers a local response, is generally removed before it can diffuse to distant parts of the cell.
TABLE 12-5 Some Proteins Regulated by and Calmodulin
Adenylyl cyclase (brain)
/calmodulin-dependent protein kinases (CaM kinases I to IV)
-dependent channel (Paramecium)
-release channel of sarcoplasmic reticulum
Calcineurin (phosphoprotein phosphatase 2B)
cAMP phosphodiesterase
cAMP-gated olfactory channel
cGMP-gated , channels (rod and cone cells)
Glutamate decarboxylase
Myosin light-chain kinases
kinase
Nitric oxide synthase
Phosphatidylinositol 3-kinase
Plasma membrane ATPase ( pump)
RNA helicase (p68)
Commonly, level does not simply rise and then fall, but rather oscillates with a period of a few seconds (Fig. 12-18) — even when the extracellular concentration of the triggering hormone remains constant. The mechanism underlying oscillations presumably entails feedback regulation by on some part of the -release process. Whatever the mechanism, the effect is that one kind of signal (hormone concentration, for example) is converted into another (frequency and amplitude of intracellular “spikes”). The signal diminishes as diffuses away from the initial source (the channel), is sequestered in the ER, or is pumped out of the cell.
FIGURE 12-18 Triggering of oscillations in intracellular by extracellular signals. (a) The dye fura, which undergoes fluorescence changes when it binds , can be used with fluorescence microscopy to measure the instantaneous in cells. The color scale relates fluorescence intensity to . Here, thymus cells have been stimulated with extracellular ATP, which raises their internal . The cells are heterogeneous in their responses: some have high intracellular (red), others have much lower (blue). (b) When such a probe is used in a single hepatocyte, the agonist norepinephrine (added at the arrow) causes oscillations of from 200 to 500 nm. Similar oscillations are induced in other cell types by other extracellular signals. [(a) Courtesy Michael D. Cahalan, University of California, Irvine, Department of Physiology and Biophysics. (b) Data from T. A. Rooney et al., J. Biol. Chem. 264:17,131, 1989.]
There is significant cross talk between the and cAMP signaling systems. In some tissues, both the enzyme that produces cAMP (adenylyl cyclase) and the enzyme that degrades cAMP (phosphodiesterase) are stimulated by . Temporal and spatial changes in can therefore produce transient, localized changes in [cAMP]. We have noted already that PKA, the enzyme that responds to cAMP, is often part of a highly localized supramolecular complex assembled on scaffold proteins such as AKAPs. This subcellular localization of target enzymes, combined with temporal and spatial gradients in and [cAMP], allows a cell to respond to one or several signals with subtly nuanced metabolic changes.
SUMMARY 12.2 G Protein–Coupled Receptors and Second Messengers
G protein–coupled receptors (GPCRs) have seven transmembrane helices and act through heterotrimeric G proteins. Ligand binding activates the G protein, which then stimulates or inhibits the activity of an effector enzyme, changing the local concentration of its second-messenger product cAMP.
Epinephrine, acting through its GPCR and the protein, stimulates adenylyl cyclase, which produces cAMP. Cyclic AMP activates protein kinase A (PKA), which then phosphorylates target proteins on a Ser or Thr residue, changing their biological activity.
To end the response to epinephrine, a phosphodiesterase breaks down cAMP, and the G protein is inactivated by its own GTPase activity. Phosphoprotein phosphatases reverse the effects of PKA.
When the epinephrine signal persists, β-adrenergic receptor–specific protein kinase phosphorylates the GPCR, creating a binding site for the protein β-arrestin, which prevents interaction between the GPCR and its G protein. Arrestin triggers desensitization by movement of the receptor into intracellular vesicles.
GPCRs typically have their effects through the G protein-cAMP-PKA pathway; some act through , raising [cAMP], others through , lowering [cAMP]. Adaptor proteins such as AKAPs tether PKA and limit its area of action and its target proteins.
Trimeric G proteins coupled to GPCRs are activated when their bound GDP is exchanged for GTP, and they remain active until their GTPase converts bound GTP to GDP. Their GTPase activity is modulated by GTPase activator proteins (GAPs) and regulators of G-protein signaling (RGSs).
Monomeric (small) G proteins such as Ras, Rab, and Ran also serve as self-limiting switches. Defective G-protein signaling is common in individuals with some types of cancer.
Some GPCRs are coupled to a G protein that acts through a plasma membrane phospholipase C that cleaves to diacylglycerol and . By opening channels in the endoplasmic reticulum, raises cytosolic . Diacylglycerol and act together to activate protein kinase C, which phosphorylates and changes the activity of specific cellular proteins.
Cellular also regulates (often through calmodulin) many other enzymes and proteins involved in secretion, cytoskeletal rearrangements, or contraction. Many of the target enzymes are in the family of -activated protein kinases (PKCs).
 Three essential components define signal transduction through GPCRs: a plasma membrane receptor with seven transmembrane helical segments, a G protein that cycles between active (guanosine triphosphate (GTP)-bound) and inactive (guanosine diphosphate (GDP)-bound) forms, and an effector enzyme (or ion channel) in the plasma membrane that is regulated by the activated G protein. An extracellular signal such as a hormone, growth factor, or neurotransmitter is the “first messenger” that activates a receptor from outside the cell. Ligand binding to the receptor forces an allosteric transition that allows the receptor to interact with a G protein, causing it to exchange its bound GDP for a GTP from the cytosol. The G protein then dissociates from the activated receptor and binds to the nearby effector enzyme, altering its activity. The effector enzyme then causes a change in the cytosolic concentration of a low molecular weight metabolite (such as ,-cyclic AMP) or inorganic ion , which acts as a
Three essential components define signal transduction through GPCRs: a plasma membrane receptor with seven transmembrane helical segments, a G protein that cycles between active (guanosine triphosphate (GTP)-bound) and inactive (guanosine diphosphate (GDP)-bound) forms, and an effector enzyme (or ion channel) in the plasma membrane that is regulated by the activated G protein. An extracellular signal such as a hormone, growth factor, or neurotransmitter is the “first messenger” that activates a receptor from outside the cell. Ligand binding to the receptor forces an allosteric transition that allows the receptor to interact with a G protein, causing it to exchange its bound GDP for a GTP from the cytosol. The G protein then dissociates from the activated receptor and binds to the nearby effector enzyme, altering its activity. The effector enzyme then causes a change in the cytosolic concentration of a low molecular weight metabolite (such as ,-cyclic AMP) or inorganic ion , which acts as a  The human genome encodes just over 800 GPCRs, about 350 for detecting hormones, growth factors, and other endogenous ligands, and up to 500 that serve as olfactory (smell) and gustatory (taste) receptors. The largest superfamily of proteins encoded in the human genome, GPCRs have been implicated in many common medical conditions, including allergies, depression, blindness, diabetes, and various cardiovascular defects. GPCR mutations are also found in 20% of all cancers. In the United States, more than a third of all pharmaceuticals on the market target a GPCR. For example, the β-adrenergic receptor, which mediates the effects of epinephrine, is the target of the “beta blockers,” prescribed for such diverse conditions as hypertension, cardiac arrhythmia, glaucoma, anxiety, and migraine headache. More than 100 of the GPCRs found in the human genome are still “orphan receptors,” meaning that their natural ligands are not yet identified, and so we know little about their biology. The β-adrenergic receptor, with well-understood biology and pharmacology, is the prototype for all GPCRs, and our discussion of signal-transducing systems begins there.
The human genome encodes just over 800 GPCRs, about 350 for detecting hormones, growth factors, and other endogenous ligands, and up to 500 that serve as olfactory (smell) and gustatory (taste) receptors. The largest superfamily of proteins encoded in the human genome, GPCRs have been implicated in many common medical conditions, including allergies, depression, blindness, diabetes, and various cardiovascular defects. GPCR mutations are also found in 20% of all cancers. In the United States, more than a third of all pharmaceuticals on the market target a GPCR. For example, the β-adrenergic receptor, which mediates the effects of epinephrine, is the target of the “beta blockers,” prescribed for such diverse conditions as hypertension, cardiac arrhythmia, glaucoma, anxiety, and migraine headache. More than 100 of the GPCRs found in the human genome are still “orphan receptors,” meaning that their natural ligands are not yet identified, and so we know little about their biology. The β-adrenergic receptor, with well-understood biology and pharmacology, is the prototype for all GPCRs, and our discussion of signal-transducing systems begins there. 
 ), allosteric transitions in the receptor and its associated G protein favor the replacement of GDP with GTP. Thus the hormone-bound GPCR acts as a
), allosteric transitions in the receptor and its associated G protein favor the replacement of GDP with GTP. Thus the hormone-bound GPCR acts as a  ) can transmit the signal from the activated receptor to the downstream effector protein, adenylyl cyclase. Because this G protein stimulates its effector, it is referred to as a
) can transmit the signal from the activated receptor to the downstream effector protein, adenylyl cyclase. Because this G protein stimulates its effector, it is referred to as a  ).
).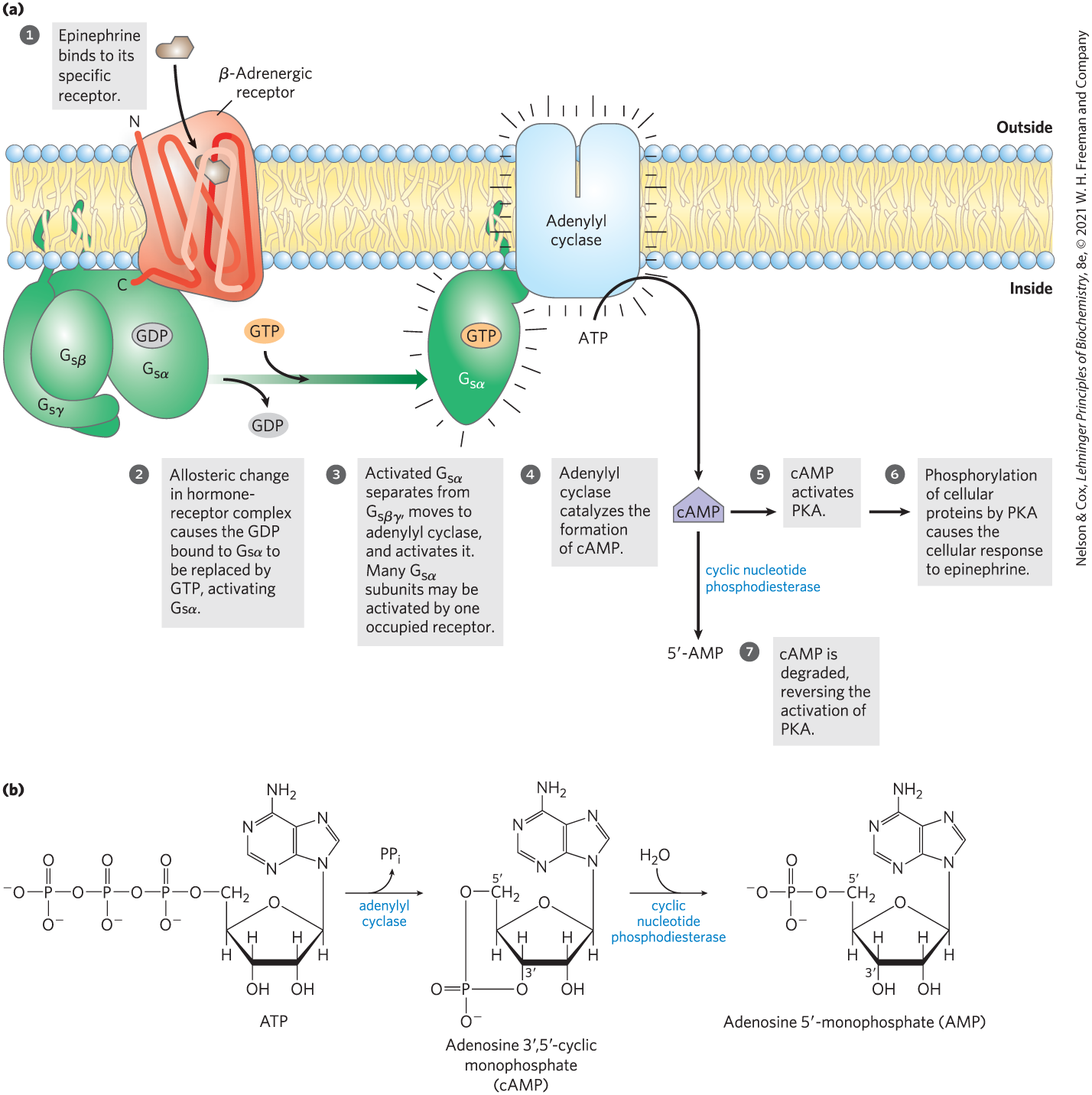
 and
and  , synthesis and hydrolysis of cAMP by adenylyl cyclase and cAMP phosphodiesterase, respectively.
, synthesis and hydrolysis of cAMP by adenylyl cyclase and cAMP phosphodiesterase, respectively.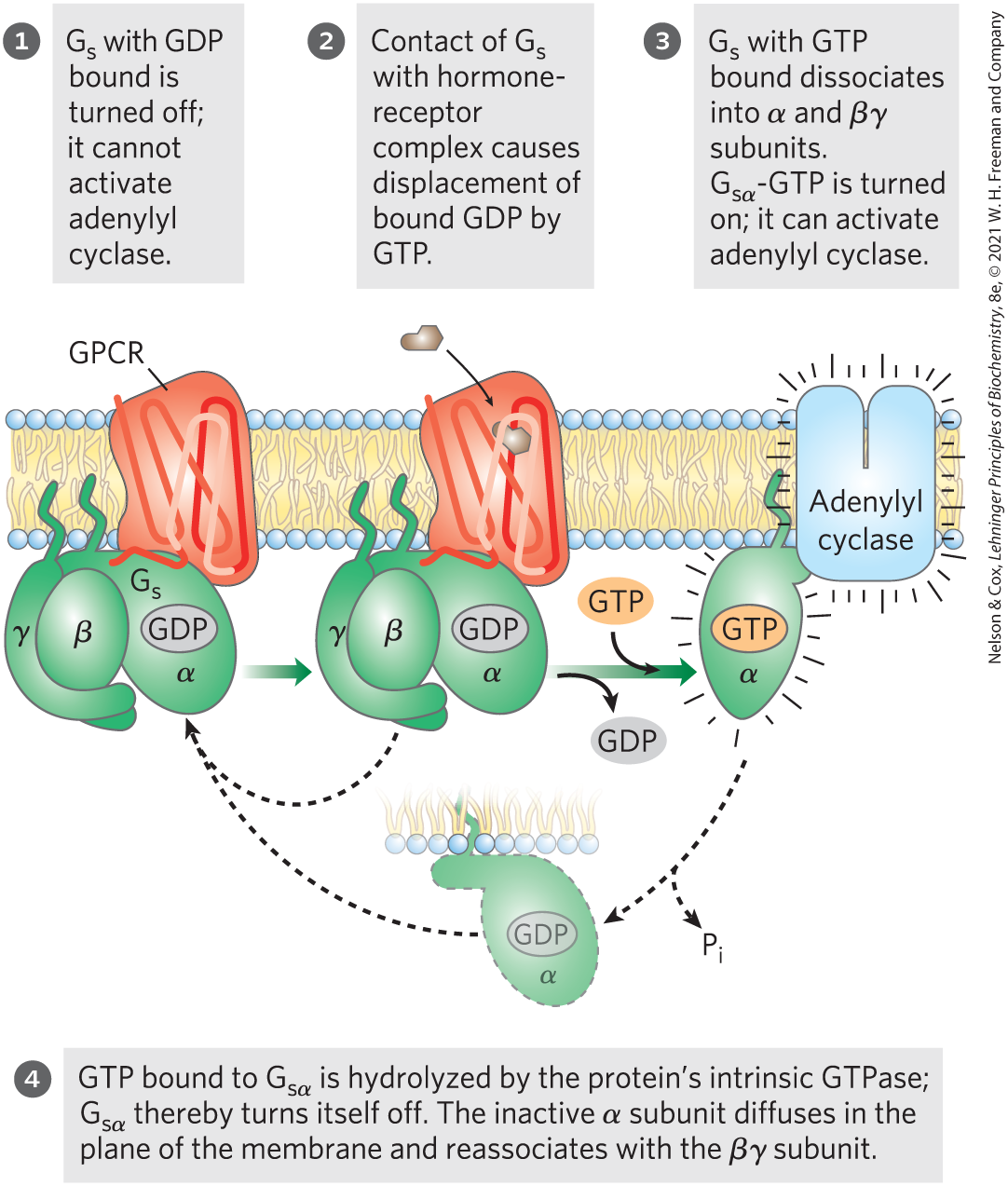
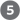 ), which catalyzes the phosphorylation of specific Ser or Thr residues of targeted proteins, such as glycogen phosphorylase b kinase. The latter enzyme is active when phosphorylated and can begin the process of mobilizing glycogen stores in muscle and liver in anticipation of the need for energy, as signaled by epinephrine.
), which catalyzes the phosphorylation of specific Ser or Thr residues of targeted proteins, such as glycogen phosphorylase b kinase. The latter enzyme is active when phosphorylated and can begin the process of mobilizing glycogen stores in muscle and liver in anticipation of the need for energy, as signaled by epinephrine. This same basic mechanism — displacement of an autoinhibitory domain — mediates the allosteric activation of many types of protein kinases by their second messengers (as in
This same basic mechanism — displacement of an autoinhibitory domain — mediates the allosteric activation of many types of protein kinases by their second messengers (as in 
 ), PKA regulates many enzymes downstream in the signaling pathway. Although these downstream targets have diverse functions, they share a region of sequence similarity around the Ser or Thr residue that undergoes phosphorylation, a sequence that marks them for regulation by PKA. The substrate-binding cleft of PKA recognizes these sequences and phosphorylates their Thr or Ser residue. Comparison of the sequences of various protein substrates for PKA has yielded the short
), PKA regulates many enzymes downstream in the signaling pathway. Although these downstream targets have diverse functions, they share a region of sequence similarity around the Ser or Thr residue that undergoes phosphorylation, a sequence that marks them for regulation by PKA. The substrate-binding cleft of PKA recognizes these sequences and phosphorylates their Thr or Ser residue. Comparison of the sequences of various protein substrates for PKA has yielded the short  As in many signaling pathways, signal transduction by the β-adrenergic receptor and adenylyl cyclase entails several steps that amplify the original hormone signal (
As in many signaling pathways, signal transduction by the β-adrenergic receptor and adenylyl cyclase entails several steps that amplify the original hormone signal (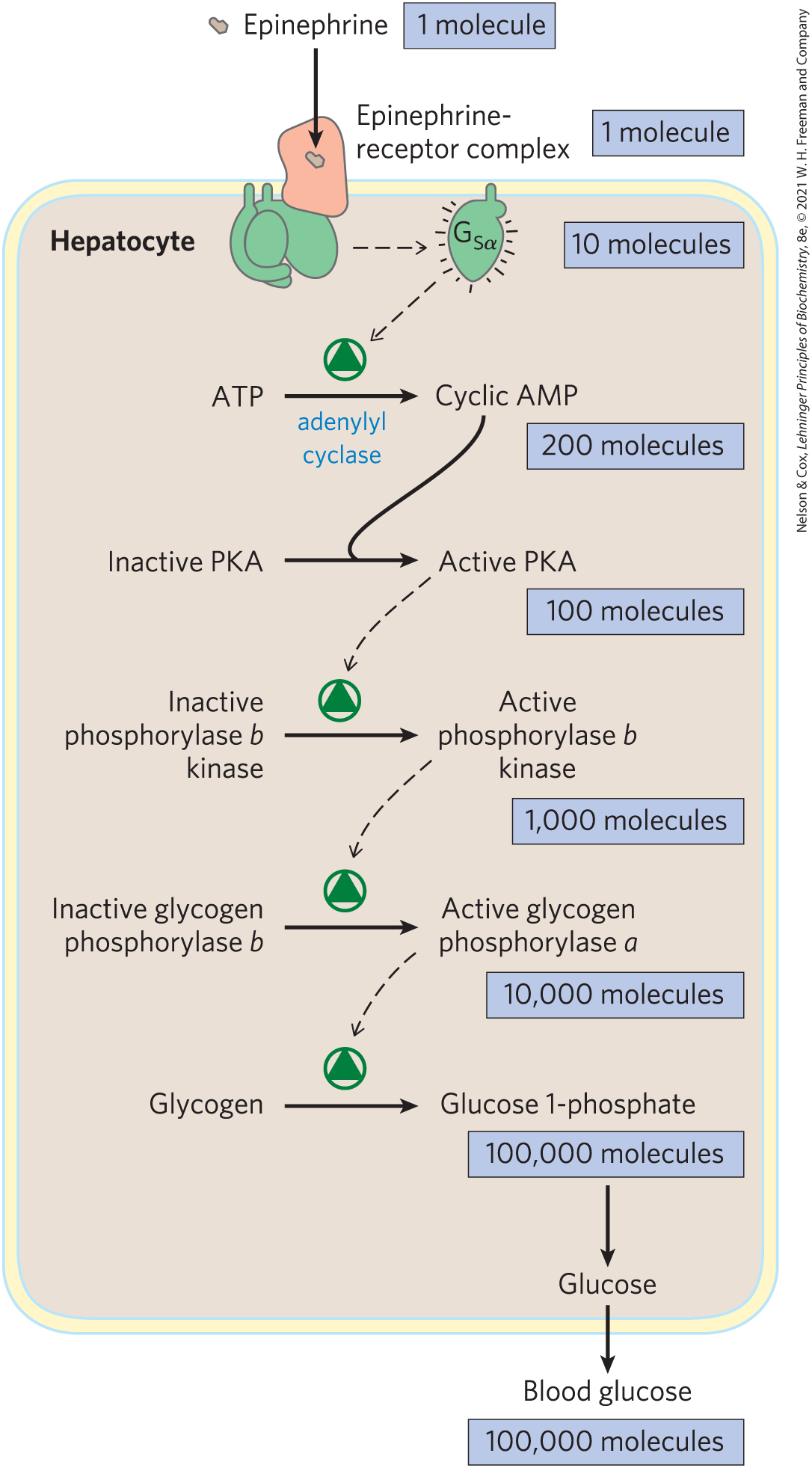
 Epinephrine is just one of many hormones, growth factors, and other regulatory molecules that act by changing the intracellular [cAMP] and thus the activity of PKA.
Epinephrine is just one of many hormones, growth factors, and other regulatory molecules that act by changing the intracellular [cAMP] and thus the activity of PKA. 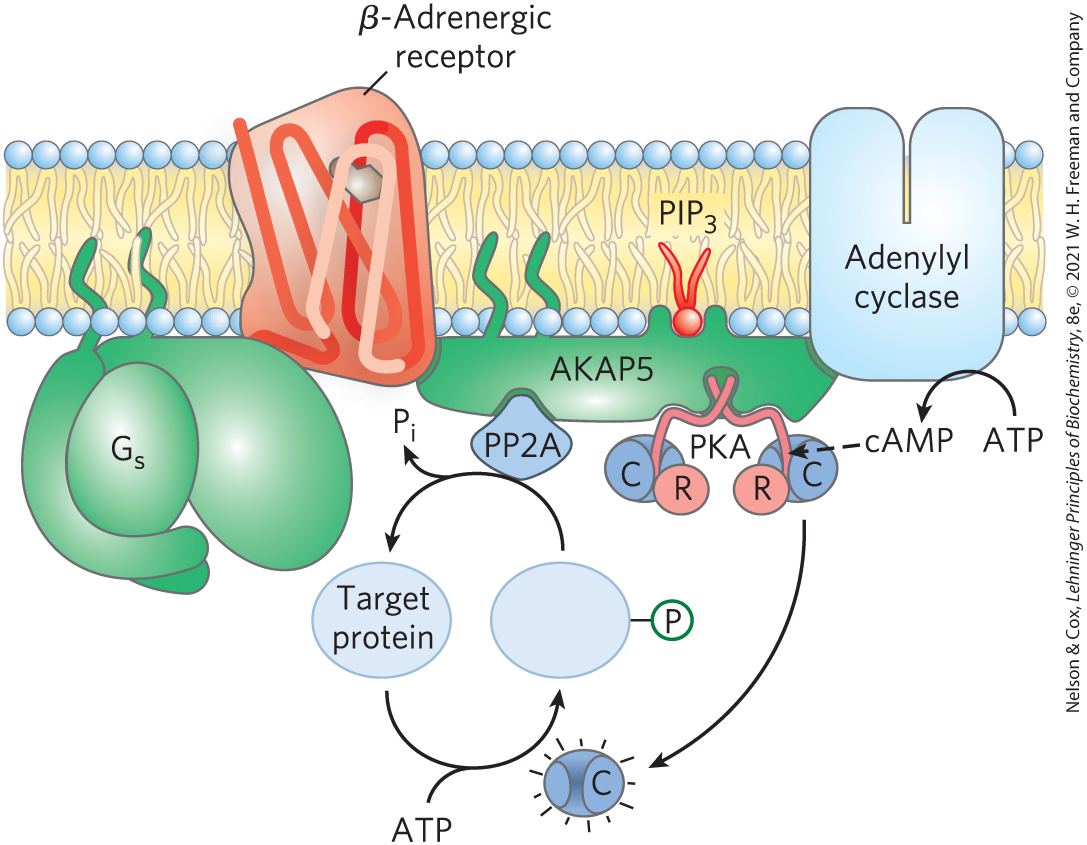
 Because G proteins play crucial roles in so many signaling processes, it is not surprising that defects in G proteins lead to a variety of diseases. In about 25% of all human cancers (and in a much higher proportion of certain types of cancer), a mutation in a Ras protein — typically in one of the critical residues around the GTP-binding site or in the P loop — virtually eliminates its GTPase activity. Once activated by GTP binding, this Ras protein remains constantly active, promoting cell division in cells that should not divide. The tumor suppressor gene NF1 encodes a GAP that enhances the GTPase activity of normal Ras. Mutations in NF1 that result in a nonfunctioning GAP leave Ras with only its GTPase activity, which is very weak (that is, has a very low turnover number); once activated by GTP binding, Ras stays active for an extended period, continuing to send the signal: divide.
Because G proteins play crucial roles in so many signaling processes, it is not surprising that defects in G proteins lead to a variety of diseases. In about 25% of all human cancers (and in a much higher proportion of certain types of cancer), a mutation in a Ras protein — typically in one of the critical residues around the GTP-binding site or in the P loop — virtually eliminates its GTPase activity. Once activated by GTP binding, this Ras protein remains constantly active, promoting cell division in cells that should not divide. The tumor suppressor gene NF1 encodes a GAP that enhances the GTPase activity of normal Ras. Mutations in NF1 that result in a nonfunctioning GAP leave Ras with only its GTPase activity, which is very weak (that is, has a very low turnover number); once activated by GTP binding, Ras stays active for an extended period, continuing to send the signal: divide.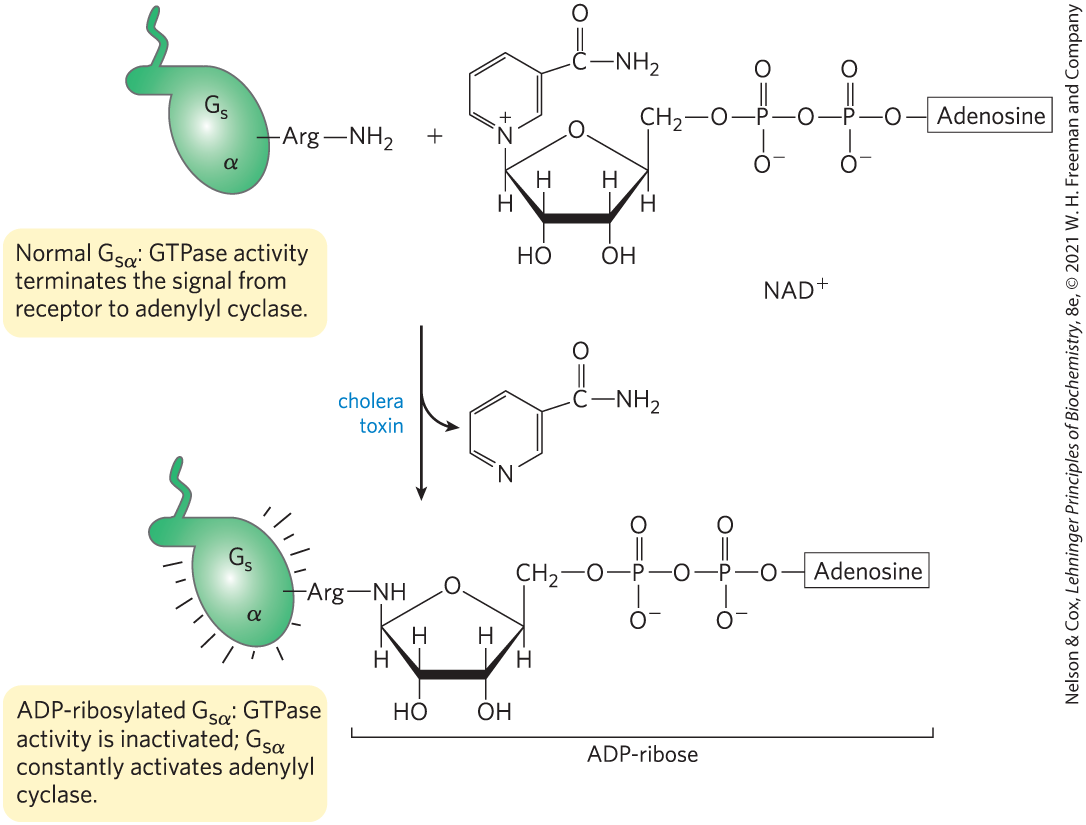
 Hormonal, neural, or other stimuli cause either an influx of into the cell through specific channels in the plasma membrane or the release of sequestered from the ER or mitochondria, in either case raising the cytosolic and triggering a cellular response.
Hormonal, neural, or other stimuli cause either an influx of into the cell through specific channels in the plasma membrane or the release of sequestered from the ER or mitochondria, in either case raising the cytosolic and triggering a cellular response.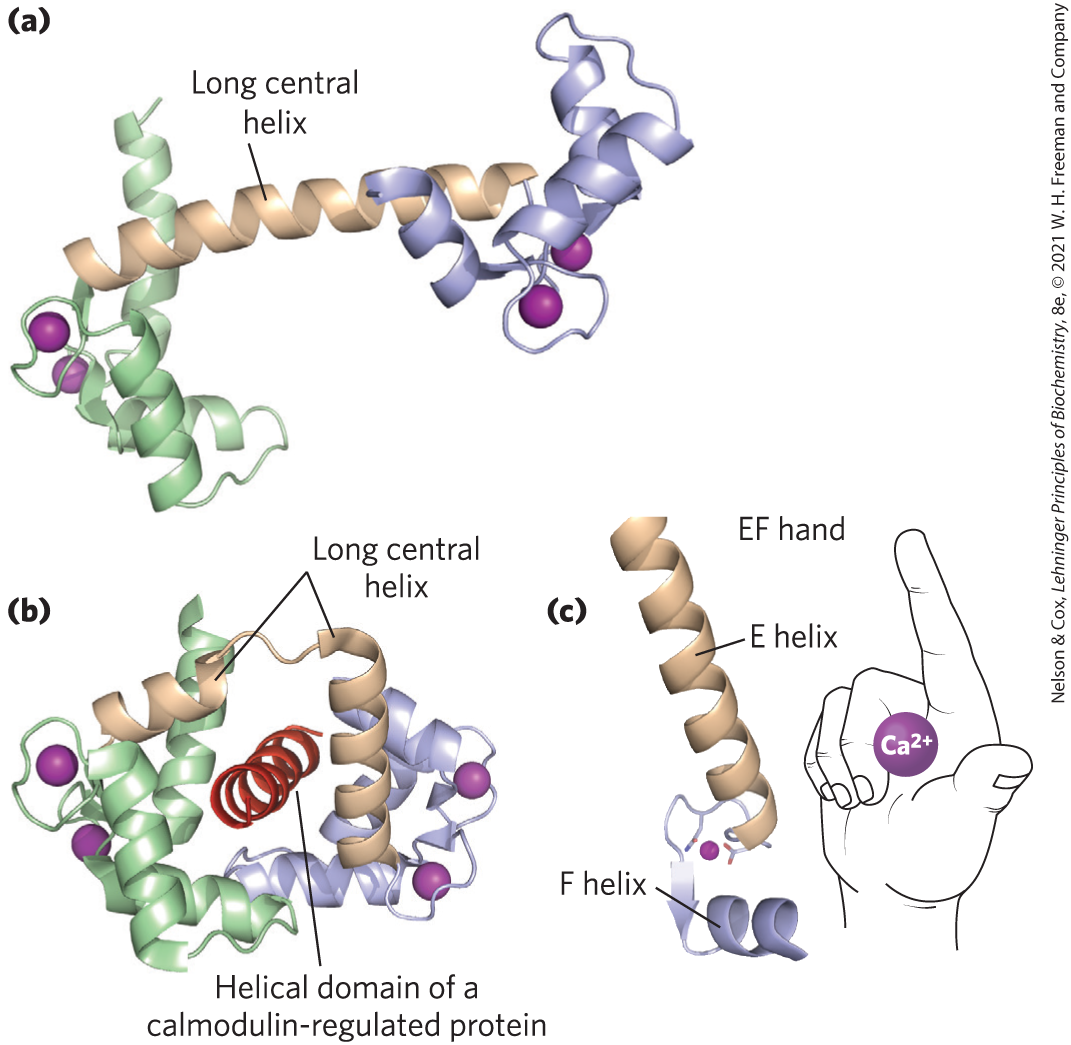
![A two-part figure shows the appearance of thymus cells stimulated with extracellular A T P, dyed with fura, and viewed using fluorescence microscopy in part a and a graph of cytosolic [C a 2 plus] (n M) plotted against time (s) in part b.](../images/8e_12_18ab_277038.png)
 G protein–coupled receptors (GPCRs) have seven transmembrane helices and act through heterotrimeric G proteins. Ligand binding activates the G protein, which then stimulates or inhibits the activity of an effector enzyme, changing the local concentration of its second-messenger product cAMP.
G protein–coupled receptors (GPCRs) have seven transmembrane helices and act through heterotrimeric G proteins. Ligand binding activates the G protein, which then stimulates or inhibits the activity of an effector enzyme, changing the local concentration of its second-messenger product cAMP.