Thus far we have focused on the general principles of catalysis and on introducing some of the kinetic parameters used to describe enzyme action. We now turn to several examples of specific enzyme reaction mechanisms.
To understand the complete mechanism of action of a purified enzyme, we need to identify all substrates, cofactors, products, and regulators. We also need to know (1) the temporal sequence in which enzyme-bound reaction intermediates form, (2) the structure of each intermediate and each transition state, (3) the rates of interconversion between intermediates, (4) the structural relationship of the enzyme to each intermediate, and (5) the energy that all reacting and interacting groups contribute to the intermediate complexes and transition states. There are still only a few enzymes for which we have an understanding that meets all these requirements.
Here we present the mechanisms for three enzymes: chymotrypsin, hexokinase, and enolase. These examples are not intended to cover all possible classes of enzyme chemistry. They have been chosen in part because they are among the best-understood enzymes and in part because they clearly illustrate some general principles outlined in this chapter. We present the chymotrypsin example in order to review some of the conventions used to depict enzyme mechanisms. Much mechanistic detail and experimental evidence is necessarily omitted; no one book could completely document the rich experimental history of these enzymes. In addition, we consider only briefly the special contribution of coenzymes to the catalytic activity of many enzymes. The function of coenzymes is chemically varied, and we describe each coenzyme in detail as it is encountered in our discussion of metabolism in Part II of this book.
The Chymotrypsin Mechanism Involves Acylation and Deacylation of a Ser Residue
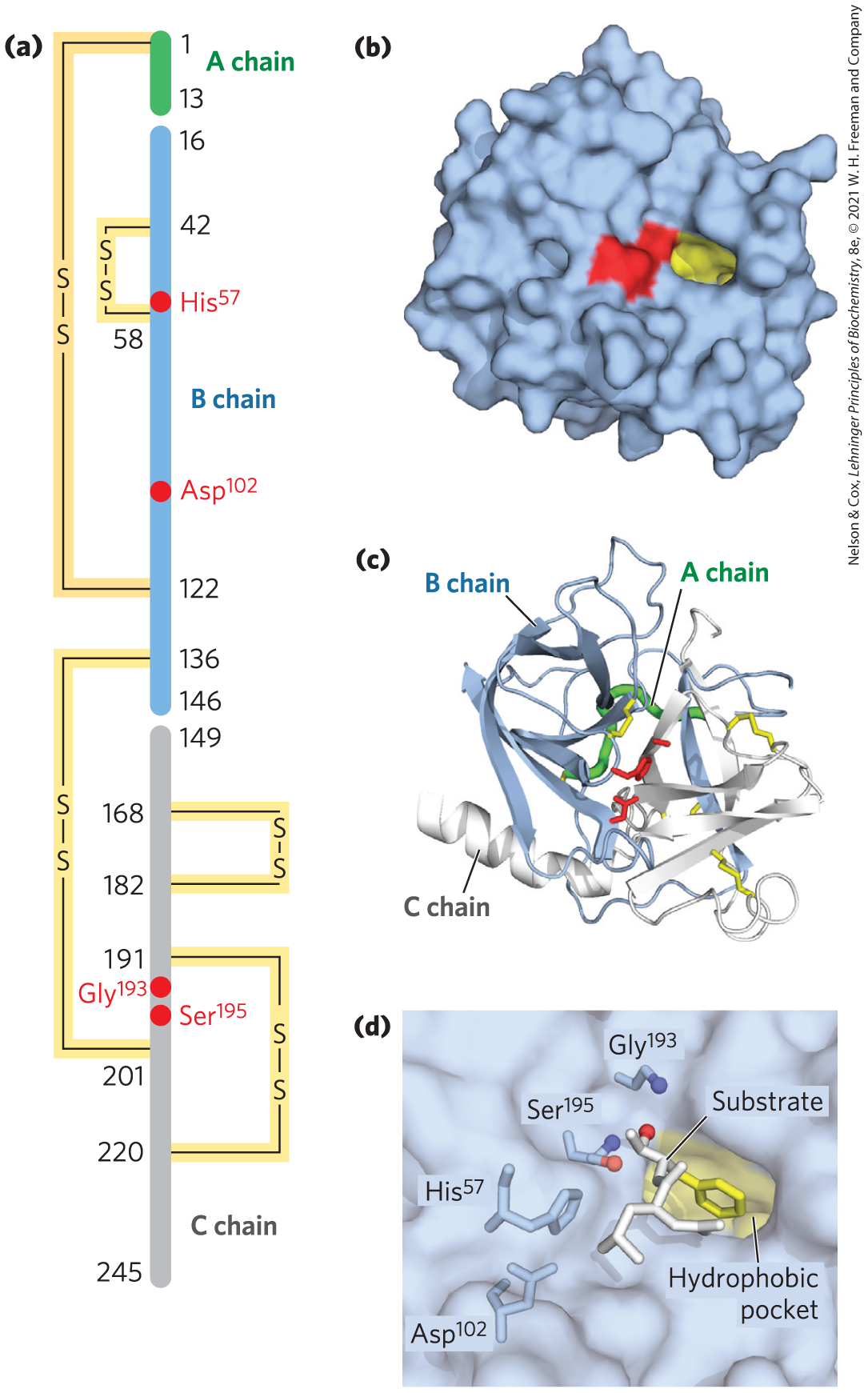
Bovine pancreatic chymotrypsin is a protease, an enzyme that catalyzes the hydrolytic cleavage of peptide bonds. This protease is specific for peptide bonds adjacent to aromatic amino acid residues (Trp, Phe, Tyr). The three-dimensional structure of chymotrypsin is shown in Figure 6-24, with functional groups in the active site emphasized. The reaction catalyzed by this enzyme illustrates the principle of transition-state stabilization and also provides a classic example of general acid-base catalysis and covalent catalysis.
FIGURE 6-24 Structure of chymotrypsin. (a) A representation of primary structure, showing disulfide bonds and the amino acid residues crucial to catalysis. The protein consists of three polypeptide chains linked by disulfide bonds. (The numbering of residues in chymotrypsin, with “missing” residues 14, 15, 147, and 148, is explained in Fig. 6-42.) The active-site amino acid residues are grouped together in the three-dimensional structure. (b) A depiction of the enzyme emphasizing its surface. The hydrophobic pocket in which the aromatic amino acid side chain of the substrate is bound is shown in yellow. Key active-site residues, including , , and , are red. The roles of these residues in catalysis are illustrated in Figure 6-27. (c) The polypeptide backbone as a ribbon structure. Disulfide bonds are yellow; the three chains are colored as in part (a). (d) A close-up of the active site with a substrate (white and yellow) bound. The hydroxyl of attacks the carbonyl group of the substrate (the oxygens are red); the developing negative charge on the oxygen is stabilized by the oxyanion hole (amide nitrogens from and , in blue), as explained in Figure 6-27. The aromatic amino acid side chain of the substrate (yellow) sits in the hydrophobic pocket. The amide nitrogen of the peptide bond to be cleaved (protruding toward the viewer and projecting the path of the rest of the substrate polypeptide chain) is shown in white. [(b, c, d) Data from PDB ID 7GCH, K. Brady et al., Biochemistry 29:7600, 1990.]
Chymotrypsin enhances the rate of peptide bond hydrolysis by a factor of at least . It does not catalyze a direct attack of water on the peptide bond; instead, a transient covalent acyl-enzyme intermediate is formed. The reaction thus has two distinct phases. In the acylation phase, the peptide bond is cleaved and an ester linkage is formed between the peptide carbonyl carbon and the enzyme. In the deacylation phase, the ester linkage is hydrolyzed and the nonacylated enzyme is regenerated.
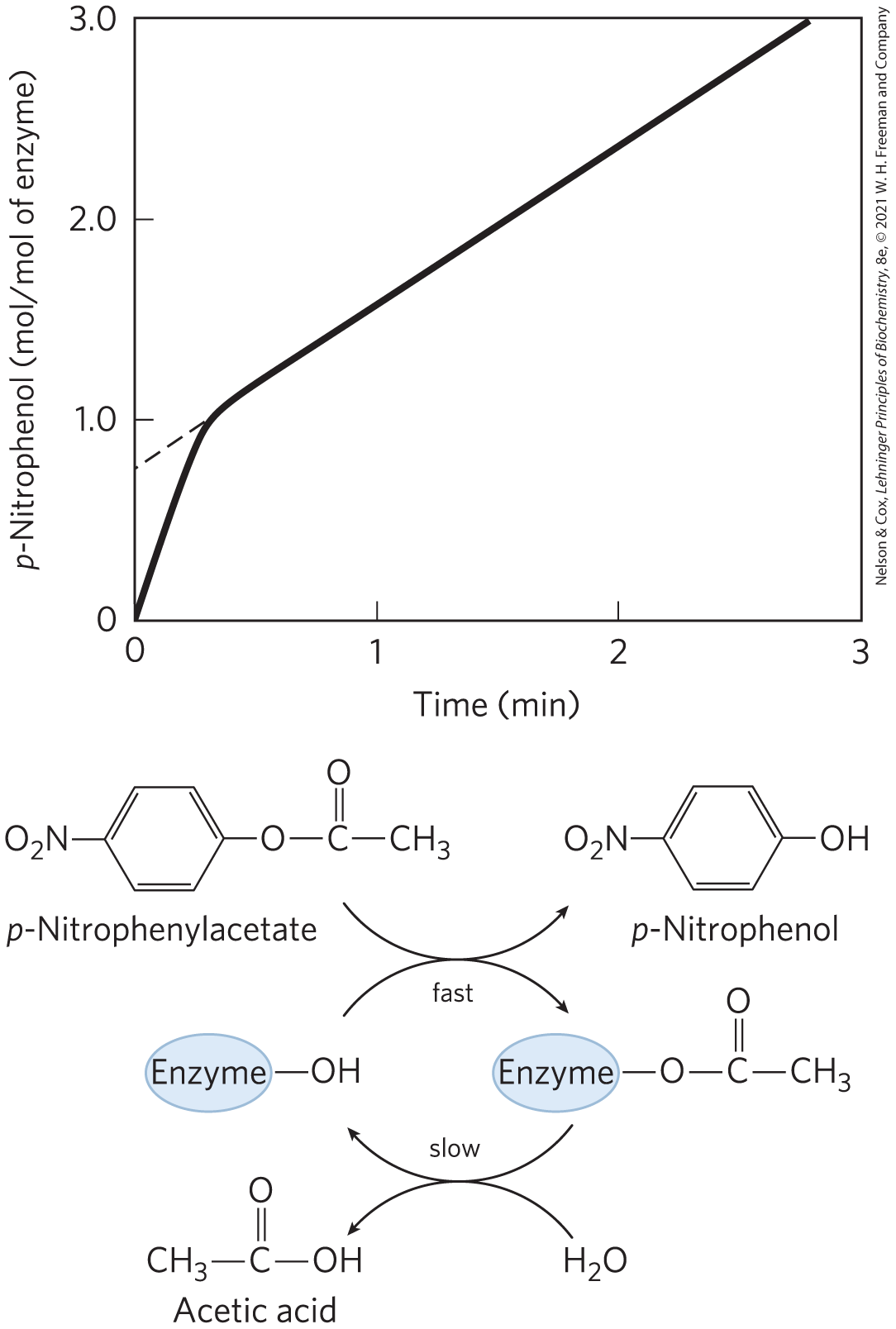
The first evidence for a covalent acyl-enzyme intermediate came from a classic application of pre–steady state kinetics. In addition to its action on polypeptides, chymotrypsin catalyzes the hydrolysis of small esters and amides. These reactions are much slower than hydrolysis of peptides because less binding energy is available with smaller substrates (the pre–steady state is also correspondingly longer), thus simplifying the analysis of the resulting reactions. In their investigations in 1954, B. S. Hartley and B. A. Kilby found that chymotrypsin hydrolysis of the ester p-nitrophenylacetate, as measured by release of p-nitrophenol, proceeds with a rapid burst before leveling off to a slower rate (Fig. 6-25). By extrapolating back to zero time, they concluded that the burst phase corresponded to the release of just under one molecule of p-nitrophenol for every enzyme molecule present (a small fraction of their enzyme molecules were inactive). Recall from Figure 6-18 that a burst implies that the rate-limiting step of catalysis occurs after release of the product being monitored. Hartley and Kilby interpreted the burst to reflect a rapid release of p-nitrophenol during a rapid acylation of all the enzyme molecules. They suggested that turnover of the enzyme was limited by a subsequent, slower deacylation step. Later work substantiated their hypothesis. The observation of a burst phase provides yet another example of the use of kinetics to break down a reaction into its constituent steps.
FIGURE 6-25 Pre–steady state kinetic evidence for an acyl-enzyme intermediate. The hydrolysis of p-nitrophenylacetate by chymotrypsin is measured by release of p-nitrophenol (a colored product). Initially, there is a rapid burst of p-nitrophenol release nearly stoichiometric with the amount of enzyme present. This reflects the fast acylation phase of the reaction. The subsequent rate is slower, because enzyme turnover is limited by the rate of the slower deacylation phase.
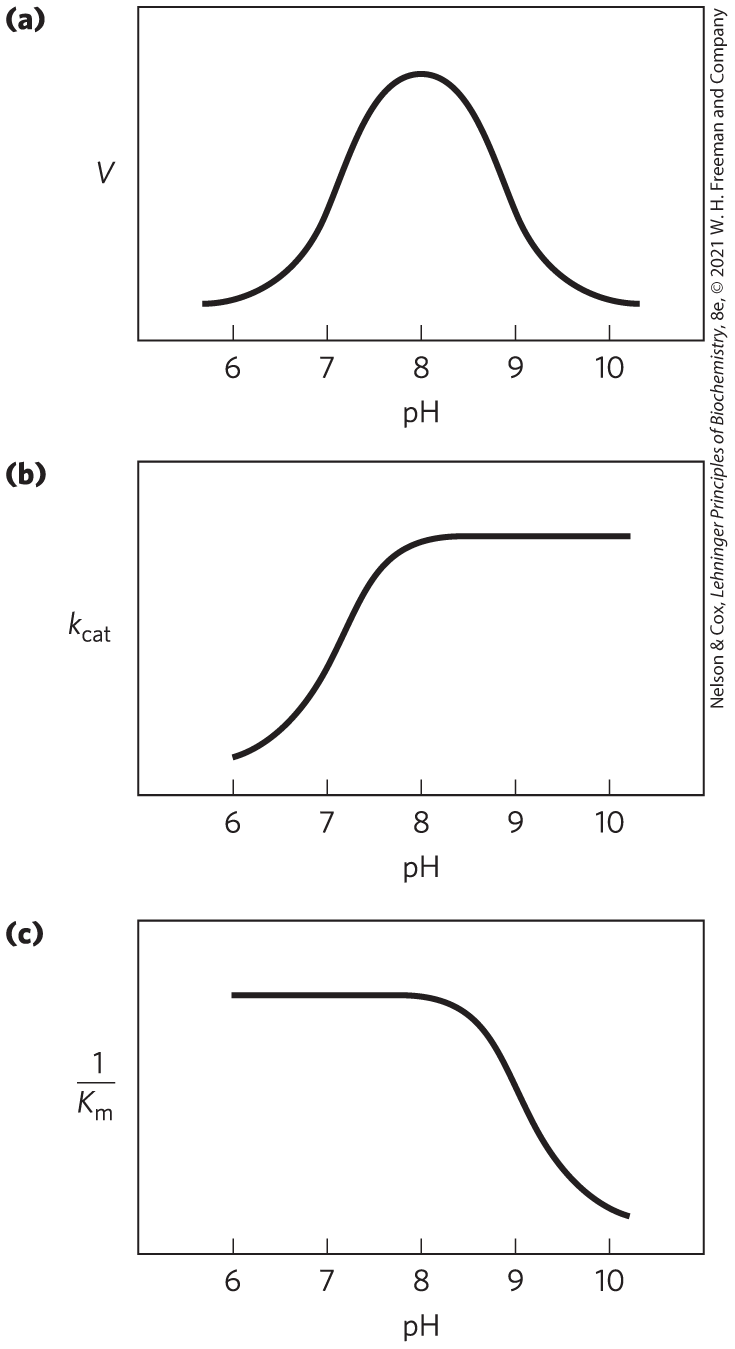
Researchers have discovered additional features of the chymotrypsin mechanism by analyzing the dependence of the reaction on pH. The rate of chymotrypsin-catalyzed cleavage generally exhibits a bell-shaped pH-rate profile (Fig. 6-26). The rates plotted in Figure 6-26a are obtained at low (subsaturating) substrate concentrations and therefore represent (see Eqn 6-30, p. 193). A more complete analysis of the rates at different substrate concentrations at each pH allows researchers to determine the individual contributions of the and terms. After obtaining the maximum rates at each pH, one can plot the alone versus pH (Fig. 6-26b); after obtaining the at each pH, researchers can then plot versus pH (Fig. 6-26c). Kinetic and structural analyses have revealed that the change in reflects the ionization state of . The decline in at low pH results from protonation of (so that it can no longer extract a proton from in the first chemical step of the reaction). This rate reduction illustrates the importance of general acid and general base catalysis in the mechanism for chymotrypsin. The changes in the term reflect the ionization of the α-amino group of (at the amino-terminal end of one of the enzyme’s three polypeptide chains). This group forms a salt bridge to , stabilizing the active conformation of the enzyme. When this group loses its proton at high pH, the salt bridge is eliminated, and a conformational change closes the hydrophobic pocket where the aromatic amino acid side chain of the substrate inserts (Fig. 6-24). Substrates can no longer bind properly, which is measured kinetically as an increase in .
FIGURE 6-26 The pH dependence of chymotrypsin-catalyzed reactions. (a) The rates of chymotrypsin-mediated cleavage produce a bell-shaped pH-rate profile with an optimum at pH 8.0. The rate (V) plotted here is that at low substrate concentrations and thus reflects the term . The plot can be broken down to its components by using kinetic methods to determine the terms and separately at each pH. When this is done (b, c), it becomes clear that the transition just above pH 7 is due to changes in , whereas the transition above pH 8.5 is due to changes in . Kinetic and structural studies have shown that the transitions illustrated in (b) and (c) reflect the ionization states of the side chain (when substrate is not bound) and the α-amino group of (at the amino terminus of the B chain), respectively. For optimal activity, must be unprotonated and must be protonated.
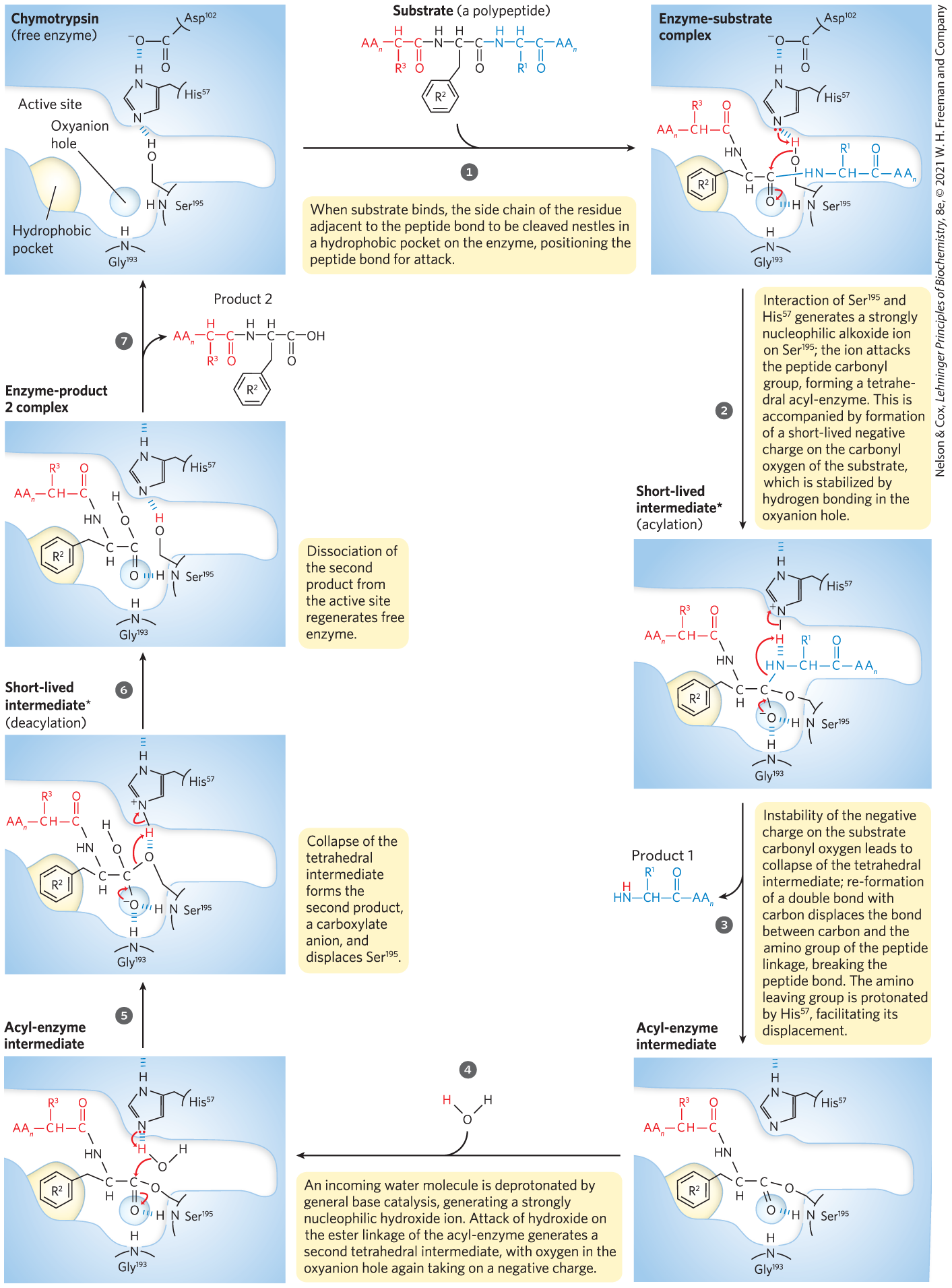
The chymotrypsin reaction is detailed in Figure 6-27. The nucleophile in the acylation phase is the oxygen of . (Proteases with a Ser residue that plays this role in reaction mechanisms are called serine proteases.) The of a Ser hydroxyl group is generally too high for the unprotonated form to be present in significant concentrations at physiological pH. However, in chymotrypsin, is linked to and in a hydrogen-bonding network referred to as the catalytic triad. When a peptide substrate binds to chymotrypsin, a subtle change in conformation compresses the hydrogen bond between and , resulting in a stronger interaction, called a low-barrier hydrogen bond. This enhanced interaction increases the of from ∼7 (for free histidine) to >12, allowing the His residue to act as an enhanced general base that can remove the proton from the hydroxyl group. Deprotonation prevents development of a highly unstable positive charge on the hydroxyl and makes the Ser side chain a stronger nucleophile. At later reaction stages, also acts as a proton donor, protonating the amino group in the displaced portion of the substrate (the leaving group).
MECHANISM FIGURE 6-27 Hydrolytic cleavage of a peptide bond by chymotrypsin. The reaction has two phases. In the acylation phase (steps to ), formation of a covalent acyl-enzyme intermediate is coupled to cleavage of the peptide bond. In the deacylation phase (steps to ), deacylation regenerates the free enzyme; this is essentially the reverse of the acylation phase, with water mirroring, in reverse, the role of the amine component of the substrate.
*The short-lived tetrahedral intermediate following step and the second tetrahedral intermediate that forms later, following step , are sometimes referred to as transition states, but this terminology can cause confusion. An intermediate is any chemical species with a finite lifetime, “finite” being defined as longer than the time required for a molecular vibration . A transition state is simply the maximum-energy species formed on the reaction coordinate, and it does not have a finite lifetime. The tetrahedral intermediates formed in the chymotrypsin reaction closely resemble, both energetically and structurally, the transition states leading to their formation and breakdown. However, the intermediate represents a committed stage of completed bond formation, whereas the transition state is part of the process of reaction. In the case of chymotrypsin, given the close relationship between the intermediate and the actual transition state, the distinction between them is routinely glossed over. Furthermore, the interaction of the negatively charged oxygen with the amide nitrogens in the oxyanion hole, often referred to as transition-state stabilization, also serves to stabilize the intermediate in this case. Not all intermediates are so short-lived that they resemble transition states. The chymotrypsin acyl-enzyme intermediate is much more stable and more readily detected and studied, and it is never confused with a transition state.
As the oxygen attacks the carbonyl group of the substrate (Fig. 6-27, step ), a very short-lived tetrahedral intermediate is formed in which the carbonyl oxygen acquires a negative charge. This charge, forming within a pocket on the enzyme called the oxyanion hole, is stabilized by hydrogen bonds contributed by the amide groups of two peptide bonds in the chymotrypsin backbone. One of these hydrogen bonds (contributed by ) is present only in this intermediate and in the transition states for its formation and breakdown; it reduces the energy required to reach these states. This is an example of the use of binding energy in catalysis through enzyme–transition state complementarity. The intermediate collapses in step , breaking the peptide bond. The amino group of the first product is protonated by , now acting as a general acid catalyst. Water is the second substrate, entering the active site in step . As water attacks the carbon in the ester linkage in step , and the resulting intermediate collapses to break the ester linkage and generate the second product in step , again acts — first as a general base to deprotonate the water, and then as a general acid to protonate the Ser oxygen as it leaves. Dissociation of the second product (step ) completes the reaction cycle.
An Understanding of Protease Mechanisms Leads to New Treatments for HIV Infection
New pharmaceutical agents are almost always designed to inhibit an enzyme. The extremely successful therapies developed to treat HIV infection provide a case in point. The human immunodeficiency virus (HIV) is the agent that causes acquired immune deficiency syndrome (AIDS). In 2018, 38 million people worldwide were living with HIV infection, with about 1.7 million new infections that year and approximately 770,000 fatalities. AIDS first surfaced as a worldwide epidemic in the 1980s; HIV was discovered soon after and was identified as a retrovirus. Retroviruses possess (1) an RNA genome and (2) an enzyme, reverse transcriptase, that is capable of using RNA to direct the synthesis of a complementary DNA. Efforts to understand HIV and develop therapies for HIV infection benefited from decades of basic research, both on enzyme mechanisms and on the properties of other retroviruses.
A retrovirus such as HIV has a relatively simple life cycle (see Fig. 26-29). Its RNA genome is converted to duplex DNA in several steps catalyzed by the reverse transcriptase (described in Chapter 26). The duplex DNA is then inserted into a chromosome in the nucleus of the host cell by the enzyme integrase (described in Chapter 25). The integrated copy of the viral genome can remain dormant indefinitely. Alternatively, it can be transcribed back into RNA, which can then be translated into proteins to construct new virus particles. Most of the viral genes are translated into large polyproteins, which are cut by an HIV protease into the individual proteins needed to make the virus (see Fig. 26-30). Only three key enzymes operate in this cycle: the reverse transcriptase, the integrase, and the protease. These enzymes thus represent the most promising drug targets.
There are four major subclasses of proteases. The serine proteases, such as chymotrypsin and trypsin, and the cysteine proteases (in which a Cys residue serves a catalytic role similar to that of Ser in the active site) form covalent enzyme-substrate complexes; the aspartyl proteases and metalloproteases do not. The HIV protease is an aspartyl protease. Two active-site Asp residues facilitate the direct attack of a water molecule on the carbonyl group of the peptide bond to be cleaved (Fig. 6-28). The initial product of this attack is an unstable tetrahedral intermediate, much like that in the chymotrypsin reaction. This intermediate is close in structure and energy to the reaction transition state. The drugs that have been developed as HIV protease inhibitors form noncovalent complexes with the enzyme, but they bind to it so tightly that they can be considered irreversible inhibitors. The tight binding is derived in part from their design as transition-state analogs. The success of these drugs makes a point worth emphasizing: the catalytic principles we have studied in this chapter are not simply abstruse ideas to be memorized — their application saves lives.
MECHANISM FIGURE 6-28 Mechanism of action of HIV protease. Two active-site Asp residues (from different subunits) act as general acid-base catalysts, facilitating the attack of water on the peptide bond. The unstable tetrahedral intermediate in the reaction pathway is shaded light red.
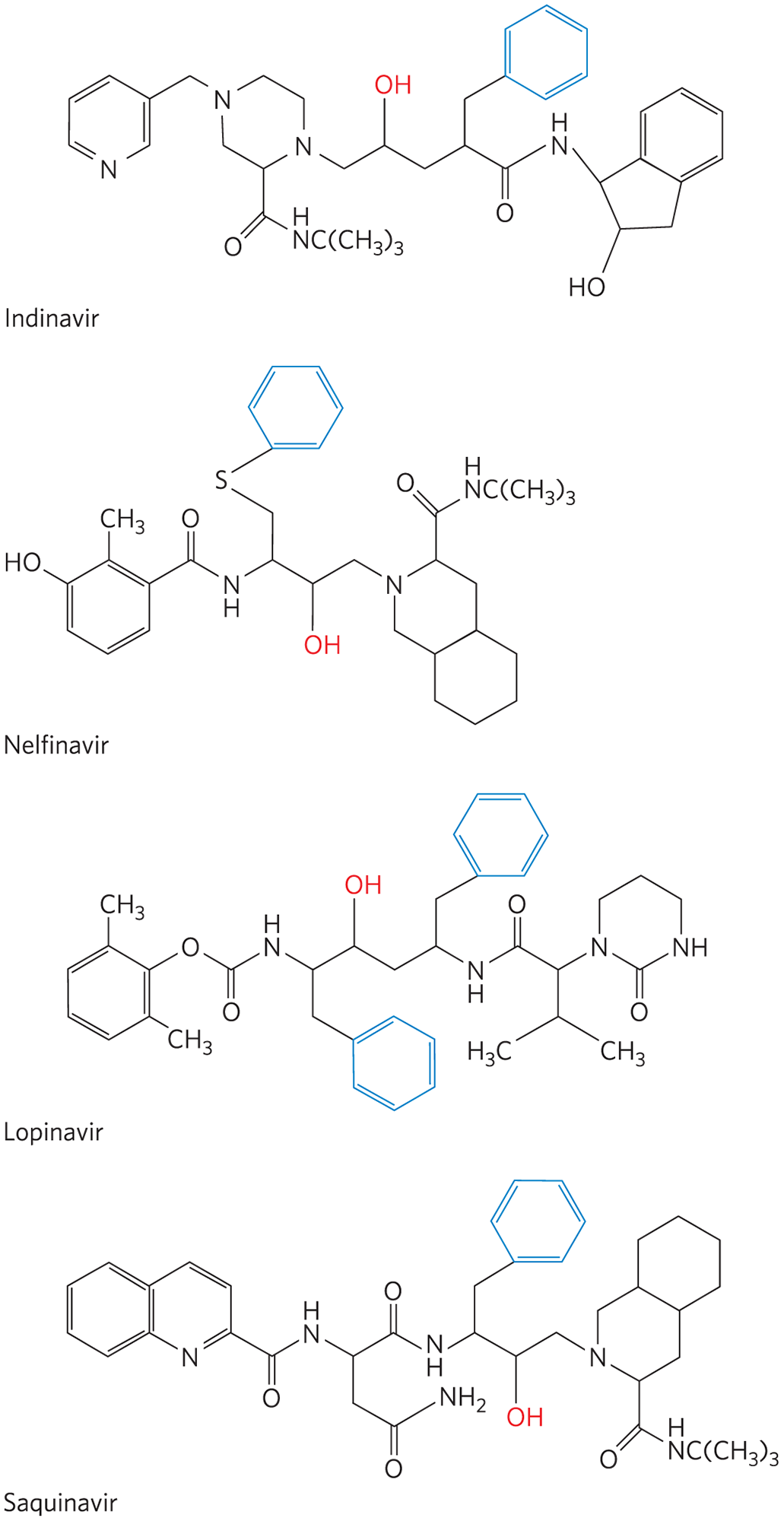
The HIV protease is most efficient at cleaving peptide bonds between Phe and Pro residues. The active site has a pocket that binds an aromatic group next to the bond to be cleaved. Several HIV protease inhibitors are shown in Figure 6-29. Although the structures appear varied, they all share a core structure: a main chain with a hydroxyl group positioned next to a branch containing a benzyl group. This arrangement targets the benzyl group to an aromatic (hydrophobic) binding pocket. The adjacent hydroxyl group mimics the negatively charged oxygen in the tetrahedral intermediate in the normal reaction, providing a transition-state analog that facilitates very tight binding. The remainder of each inhibitor structure was designed to fit into and bind to various crevices along the surface of the enzyme, enhancing overall binding. The availability of these effective drugs has vastly increased the life span and quality of life of millions of people with HIV and AIDS. In 2018, 23.3 million of the 38 million people living with HIV infection were receiving antiretroviral therapy.
FIGURE 6-29 HIV protease inhibitors. The hydroxyl group (red) acts as a transition-state analog, mimicking the oxygen of the tetrahedral intermediate. The adjacent benzyl group (blue) helps to properly position the drug in the active site.
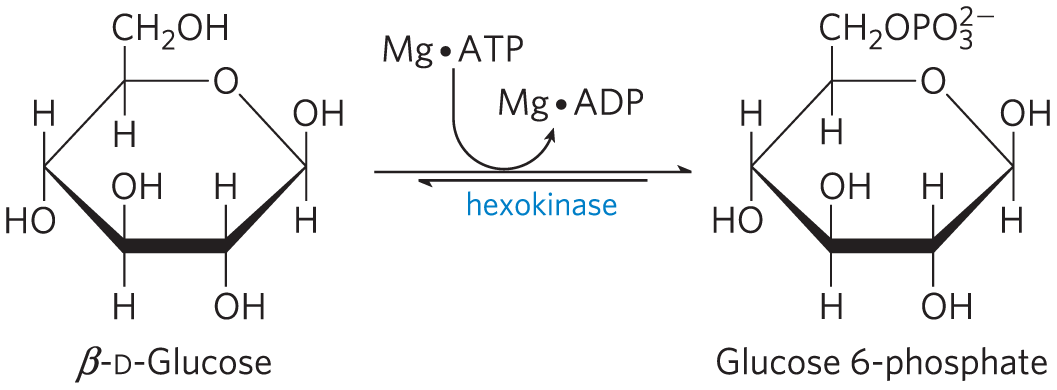
Hexokinase Undergoes Induced Fit on Substrate Binding
Yeast hexokinase is a bisubstrate enzyme that catalyzes this reversible reaction:
ATP and ADP always bind to enzymes as a complex with the metal ion .
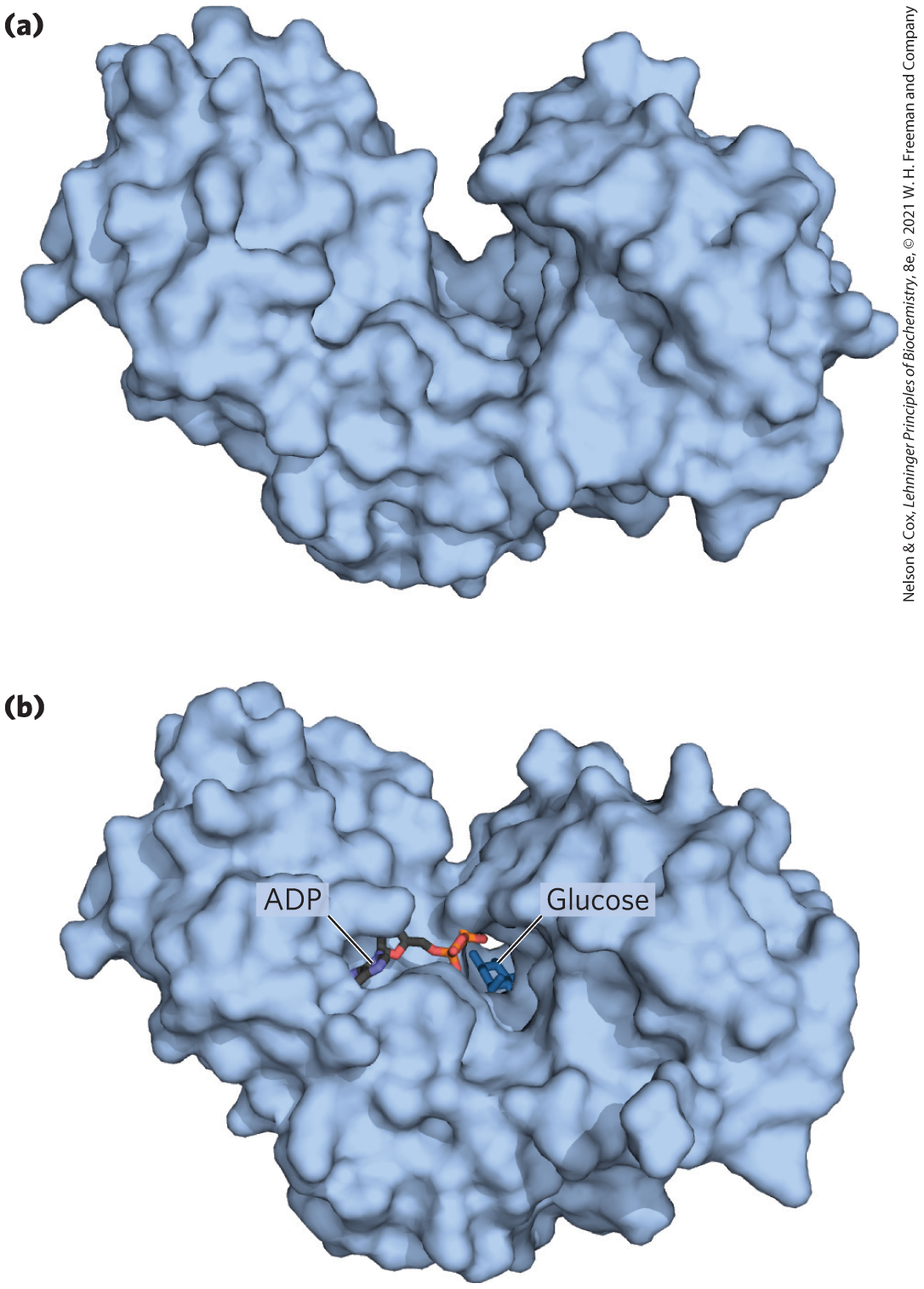
In the hexokinase reaction, the -phosphoryl of ATP is transferred to the hydroxyl at C-6 of glucose. This hydroxyl is similar in chemical reactivity to water, and water freely enters the enzyme active site. Yet hexo-kinase favors the reaction with glucose by a factor of . The enzyme can discriminate between glucose and water because of a conformational change in the enzyme when the correct substrate binds (Fig. 6-30). Hexokinase thus provides a good example of induced fit. When glucose is not present, the enzyme is in an inactive conformation, with the active-site amino acid side chains out of position for reaction. When glucose (but not water) and Mg⋅ATP bind, the binding energy derived from this interaction induces a conformational change in hexokinase to the catalytically active form.
FIGURE 6-30 Induced fit in hexokinase. (a) Hexokinase has a U-shaped structure. (b) The ends pinch toward each other in a conformational change induced by binding d-glucose. [Data from (a) PDB ID 2YHX, C. M. Anderson et al., J. Mol. Biol. 123:15, 1978. (b) PDB ID 2E2O, modeled with ADP derived from PDB ID 2E2Q, H. Nishimasu et al., J. Biol. Chem. 282:9923, 2007.]
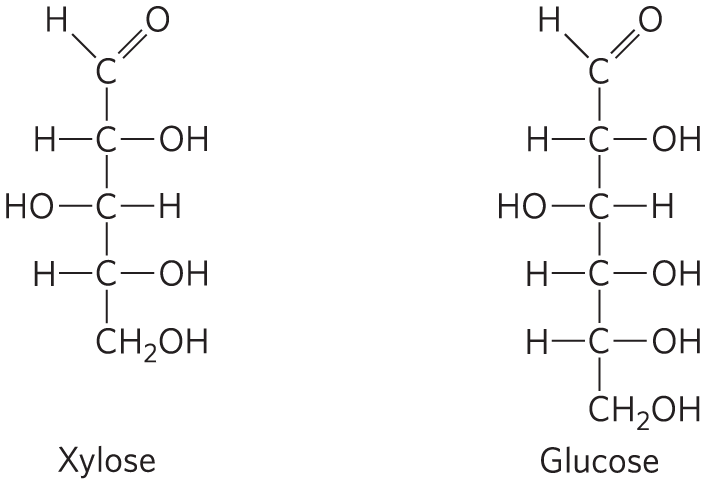
This model has been reinforced by kinetic studies. The five-carbon sugar xylose, stereochemically similar to glucose but one carbon shorter, binds to hexokinase but in a position where it cannot be phosphorylated. Nevertheless, addition of xylose to the reaction mixture increases the rate of ATP hydrolysis. Evidently, the binding of xylose is sufficient to induce a change in hexokinase to its active conformation, and the enzyme is thereby “tricked” into phosphorylating water. The hexokinase reaction also illustrates that enzyme specificity is not always a simple matter of binding one compound but not another. In the case of hexokinase, specificity is observed not in the formation of the ES complex but in the relative rates of subsequent catalytic steps. Reaction rates increase greatly in the presence of a substrate, glucose, that is able to accept a phosphoryl group.
Induced fit is only one aspect of the catalytic mechanism of hexokinase — like chymotrypsin, hexokinase uses several catalytic mechanisms. For example, the active-site amino acid residues (those brought into position by the conformational change that follows substrate binding) participate in general acid-base catalysis and transition-state stabilization.
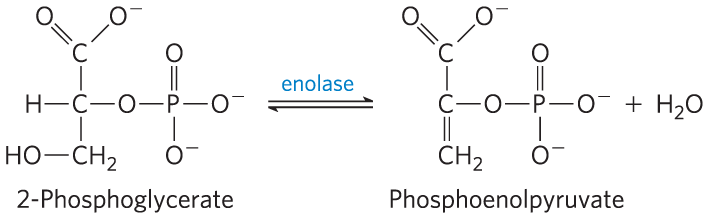
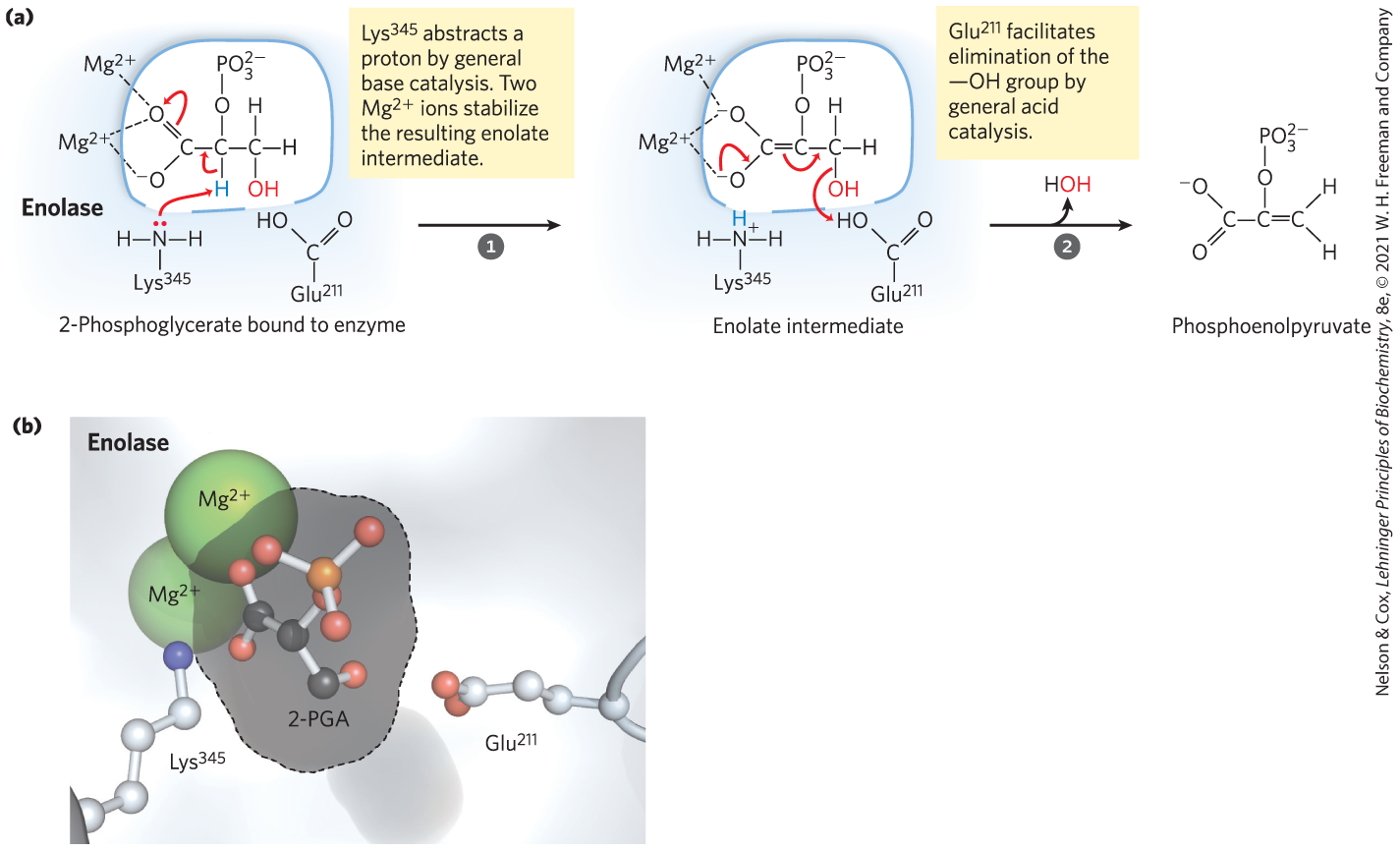
The Enolase Reaction Mechanism Requires Metal Ions
Another glycolytic enzyme, enolase, catalyzes the reversible dehydration of 2-phosphoglycerate to phosphoenolpyruvate:
The reaction provides an example of the use of an enzymatic cofactor, in this case a metal ion (another example of coenzyme function is provided in Box 6-1). Yeast enolase is a dimer with 436 amino acid residues per subunit. The enolase reaction illustrates one type of metal ion catalysis and provides an additional example of general acid-base catalysis and transition-state stabilization. The reaction occurs in two steps (Fig. 6-31a). First, acts as a general base catalyst, abstracting a proton from C-2 of 2-phosphoglycerate; then acts as a general acid catalyst, donating a proton to the leaving group. The proton at C-2 of 2-phosphoglycerate is not acidic and thus is quite resistant to its removal by . However, the electronegative oxygen atoms of the adjacent carboxyl group pull electrons away from C-2, making the attached protons somewhat more labile. In the active site, the carboxyl group of 2-phosphoglycerate undergoes strong ionic interactions with two bound ions (Fig. 6-31b), greatly enhancing the electron withdrawal by the carboxyl. Together, these effects render the C-2 protons sufficiently acidic (lowering the ) that one proton can be abstracted to initiate the reaction. As the unstable enolate intermediate is formed, the metal ions further act to shield the two negative charges (on the carboxyl oxygen atoms) that transiently exist in close proximity to each other. Hydrogen bonding to other active-site amino acid residues also contributes to the overall mechanism. The various interactions effectively stabilize both the enolate intermediate and the transition state preceding its formation.
MECHANISM FIGURE 6-31 Two-step reaction catalyzed by enolase. (a) The mechanism by which enolase converts 2-phosphoglycerate (2-PGA) to phosphoenolpyruvate. The carboxyl group of 2-PGA is coordinated by two ions at the active site. (b) The substrate, 2-PGA, in relation to the , , and in the enolase active site (gray outline). Nitrogen is shown in blue, phosphorus in orange; hydrogen atoms are not shown. [(b) Data from PDB ID 1ONE, T. M. Larsen et al., Biochemistry 35:4349, 1996.]
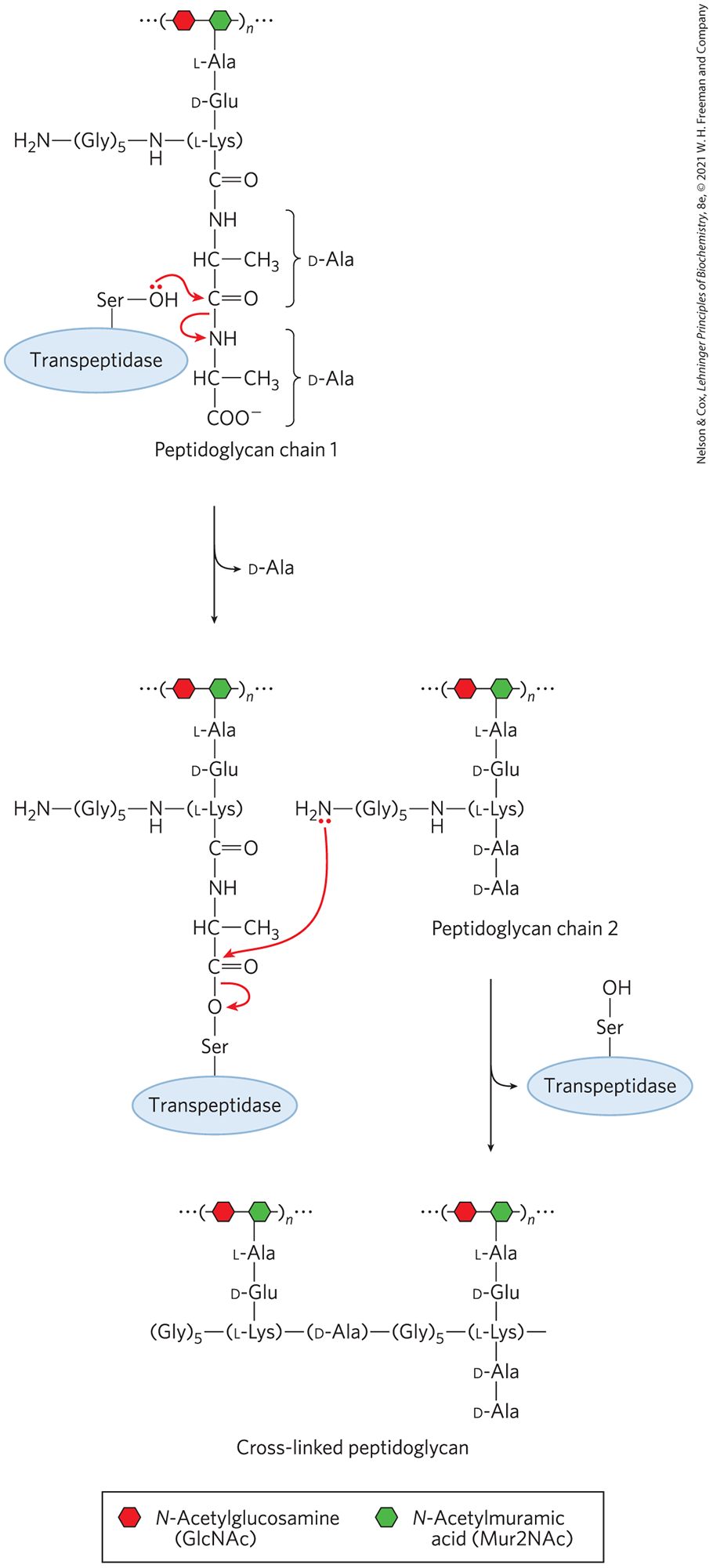
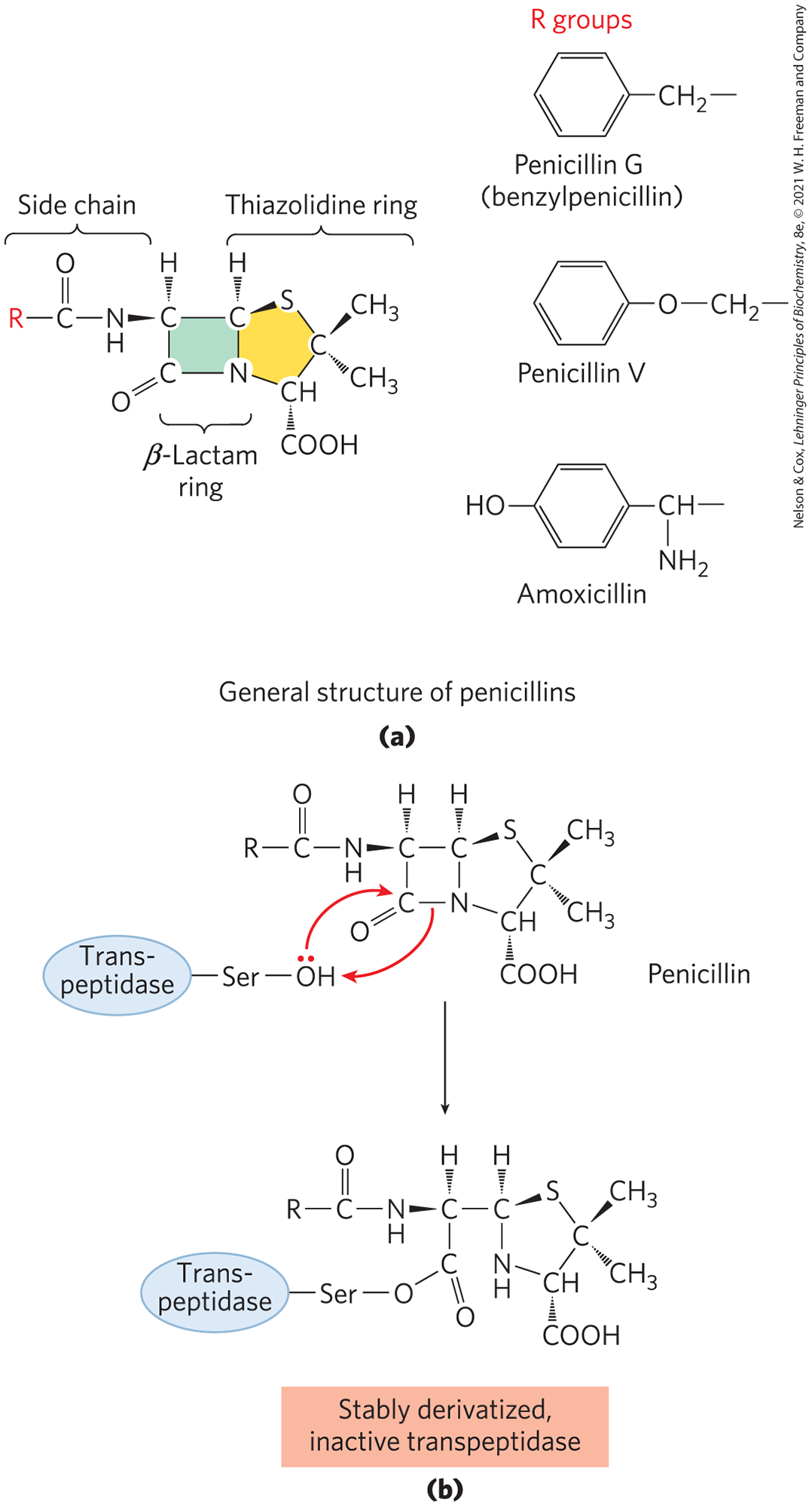
An Understanding of Enzyme Mechanism Produces Useful Antibiotics
Penicillin was discovered in 1928 by Alexander Fleming, but another 15 years passed before this relatively unstable compound was understood well enough for it to be used as a pharmaceutical agent to treat bacterial infections. Penicillin interferes with the synthesis of peptidoglycan, the major component of the rigid cell wall that protects bacteria from osmotic lysis. Peptidoglycan consists of polysaccharides and peptides cross-linked in several steps that include a transpeptidase reaction (Fig. 6-32). It is this reaction that is inhibited by penicillin and related compounds (Fig. 6-33a), all of which are irreversible inhibitors of transpeptidase. They bind to the active site of transpeptidase through a segment that mimics one conformation of the d-Ala–d-Ala segment of the peptidoglycan precursor. The peptide bond in the precursor is replaced by a highly reactive β-lactam ring in the antibiotic. When penicillin binds to the transpeptidase, an active-site Ser attacks the carbonyl of the β-lactam ring and generates a covalent adduct between penicillin and the enzyme. The leaving group remains attached, however, because it is linked by the remnant of the β-lactam ring (Fig. 6-33b). The covalent complex irreversibly inactivates the enzyme. This, in turn, blocks synthesis of the bacterial cell wall, and most bacteria die as the fragile inner membrane bursts under osmotic pressure.
FIGURE 6-32 The transpeptidase reaction. This reaction, which links two peptidoglycan precursors into a larger polymer, is facilitated by an active-site Ser and a covalent catalytic mechanism similar to that of chymotrypsin. Note that peptidoglycan is one of the few places in nature where d-amino acid residues are found. The active-site Ser attacks the carbonyl of the peptide bond between the two d-Ala residues, creating a covalent ester linkage between the substrate and the enzyme, with release of the terminal d-Ala residue. An amino group from the second peptidoglycan precursor then attacks the ester linkage, displacing the enzyme and cross-linking the two precursors.
FIGURE 6-33 Transpeptidase inhibition by β-lactam antibiotics. (a) β-Lactam antibiotics have a five-membered thiazolidine ring fused to a four-membered β-lactam ring. The latter ring is strained and includes an amide moiety that plays a critical role in the inactivation of peptidoglycan synthesis. The R group differs with the type of penicillin. Penicillin G was the first to be isolated and remains one of the most effective, but it is degraded by stomach acid and must be administered by injection. Penicillin V is nearly as effective and is acid stable, so it can be administered orally. Amoxicillin has a broad range of effectiveness, is readily administered orally, and thus is the most widely prescribed β-lactam antibiotic. (b) Attack on the amide moiety of the β-lactam ring by a transpeptidase active-site Ser results in a covalent acyl-enzyme product. This is hydrolyzed so slowly that adduct formation is practically irreversible, and the transpeptidase is inactivated.
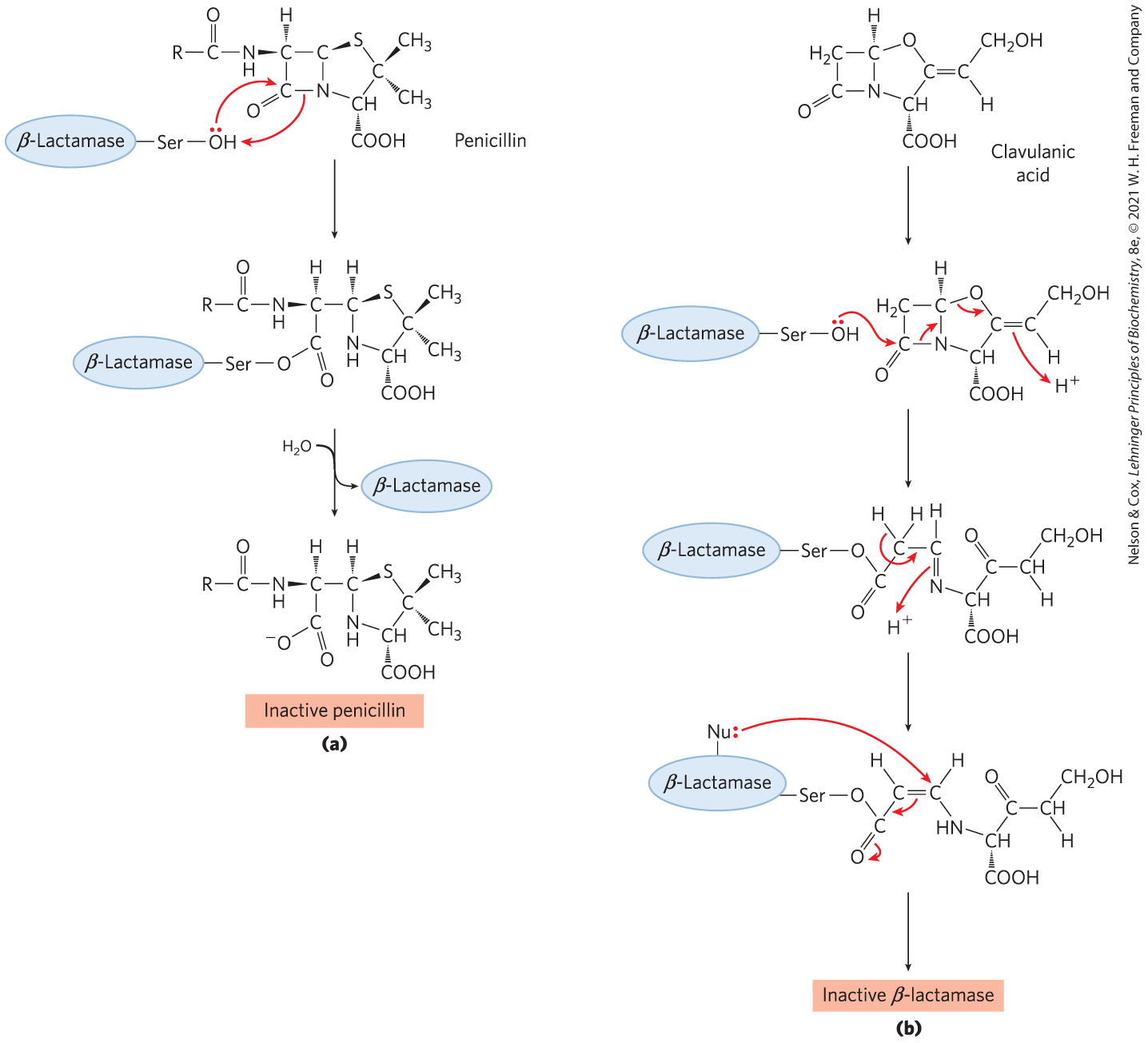
Human use of penicillin and its derivatives has led to the evolution of strains of pathogenic bacteria that express β-lactamases (Fig. 6-34a), enzymes that cleave β-lactam antibiotics, rendering them inactive. The bacteria thereby become resistant to the antibiotics. The genes for these enzymes have spread rapidly through bacterial populations under the selective pressure imposed by the use (and often overuse) of β-lactam antibiotics. Human medicine responded with the development of compounds such as clavulanic acid, a suicide inactivator, which irreversibly inactivates the β-lactamases (Fig. 6-34b). Clavulanic acid mimics the structure of a β-lactam antibiotic and forms a covalent adduct with a Ser in the β-lactamase active site. This leads to a rearrangement that creates a much more reactive derivative, which is subsequently attacked by another nucleophile in the active site to irreversibly acylate the enzyme and inactivate it. Amoxicillin and clavulanic acid are combined in a widely used pharmaceutical formulation with the trade name Augmentin. The cycle of chemical warfare between humans and bacteria continues unabated. Strains of disease-causing bacteria that are resistant to both amoxicillin and clavulanic acid have been discovered. Mutations in β-lactamase within these strains render it unreactive to clavulanic acid. The development of new antibiotics promises to be a growth industry for the foreseeable future.
FIGURE 6-34 β-Lactamases and β-lactamase inhibition. (a) β-Lactamases promote cleavage of the β-lactam ring in β-lactam antibiotics, thus inactivating them. (b) Clavulanic acid is a suicide inhibitor, making use of the normal chemical mechanism of β-lactamases to create a reactive species at the active site. This reactive species is attacked by a nucleophilic group (Nu:) in the active site to irreversibly acylate the enzyme.
SUMMARY 6.4 Examples of Enzymatic Reactions
Chymotrypsin is a serine protease with a well-understood mechanism, featuring general acid-base catalysis, covalent catalysis, and transition-state stabilization.
Hexokinase provides an excellent example of induced fit as a means of using substrate binding energy.
The enolase reaction proceeds via metal ion catalysis.
Understanding enzyme mechanism allows for the development of drugs to inhibit enzyme action.

 to
to  ), formation of a covalent acyl-enzyme intermediate is coupled to cleavage of the peptide bond. In the deacylation phase (steps
), formation of a covalent acyl-enzyme intermediate is coupled to cleavage of the peptide bond. In the deacylation phase (steps  to
to  ), deacylation regenerates the free enzyme; this is essentially the reverse of the acylation phase, with water mirroring, in reverse, the role of the amine component of the substrate.
), deacylation regenerates the free enzyme; this is essentially the reverse of the acylation phase, with water mirroring, in reverse, the role of the amine component of the substrate. and the second tetrahedral intermediate that forms later, following step
and the second tetrahedral intermediate that forms later, following step  This is an example of the use of binding energy in catalysis through enzyme–transition state complementarity. The intermediate collapses in step
This is an example of the use of binding energy in catalysis through enzyme–transition state complementarity. The intermediate collapses in step  , breaking the peptide bond. The amino group of the first product is protonated by , now acting as a general acid catalyst. Water is the second substrate, entering the active site in step
, breaking the peptide bond. The amino group of the first product is protonated by , now acting as a general acid catalyst. Water is the second substrate, entering the active site in step  , again acts — first as a general base to deprotonate the water, and then as a general acid to protonate the Ser oxygen as it leaves. Dissociation of the second product (step
, again acts — first as a general base to deprotonate the water, and then as a general acid to protonate the Ser oxygen as it leaves. Dissociation of the second product (step  New pharmaceutical agents are almost always designed to inhibit an enzyme. The extremely successful therapies developed to treat HIV infection provide a case in point. The human immunodeficiency virus (HIV) is the agent that causes acquired immune deficiency syndrome (AIDS). In 2018, 38 million people worldwide were living with HIV infection, with about 1.7 million new infections that year and approximately 770,000 fatalities. AIDS first surfaced as a worldwide epidemic in the 1980s; HIV was discovered soon after and was identified as a
New pharmaceutical agents are almost always designed to inhibit an enzyme. The extremely successful therapies developed to treat HIV infection provide a case in point. The human immunodeficiency virus (HIV) is the agent that causes acquired immune deficiency syndrome (AIDS). In 2018, 38 million people worldwide were living with HIV infection, with about 1.7 million new infections that year and approximately 770,000 fatalities. AIDS first surfaced as a worldwide epidemic in the 1980s; HIV was discovered soon after and was identified as a 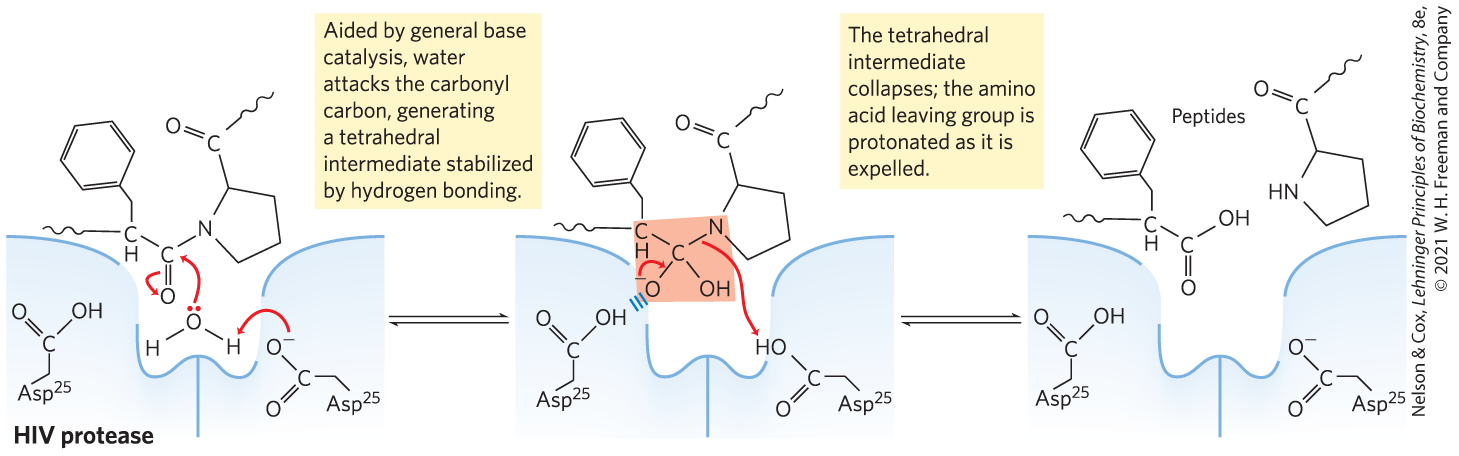

 Chymotrypsin is a serine protease with a well-understood mechanism, featuring general acid-base catalysis, covalent catalysis, and transition-state stabilization.
Chymotrypsin is a serine protease with a well-understood mechanism, featuring general acid-base catalysis, covalent catalysis, and transition-state stabilization.