4.4 Protein Denaturation and Folding
Proteins lead a surprisingly precarious existence. As we have seen, a native protein conformation is only marginally stable. In addition, most proteins must maintain conformational flexibility in order to function. The continual maintenance of the active set of cellular proteins required under a given set of conditions is called proteostasis. Cellular proteostasis requires the coordinated function of pathways for protein synthesis and folding, the refolding of proteins that are partially unfolded, and the sequestration and degradation of proteins that have been irreversibly unfolded or are no longer needed. In all cells, these networks involve hundreds of enzymes and specialized proteins.
 The continual maintenance of the active set of cellular proteins required under a given set of conditions is called
The continual maintenance of the active set of cellular proteins required under a given set of conditions is called As seen in Figure 4-23, the life of a protein encompasses much more than its synthesis and later degradation. The marginal stability of most proteins can produce a tenuous balance between folded and unfolded states. As proteins are synthesized on ribosomes (Chapter 27), they must fold into their native conformations. Sometimes this occurs spontaneously, but often it requires the assistance of specialized enzymes and complexes called chaperones, which we discuss later in the chapter. Many of these same folding helpers function to refold proteins that become transiently unfolded. Proteins that are not properly folded often have exposed hydrophobic surfaces that render them “sticky,” leading to the formation of inactive aggregates. These aggregates may lack their normal function but are not inert; their accumulation in cells lies at the heart of diseases ranging from diabetes to Parkinson disease and Alzheimer disease. Not surprisingly, all cells have elaborate pathways for recycling and/or degrading proteins that are irreversibly misfolded.
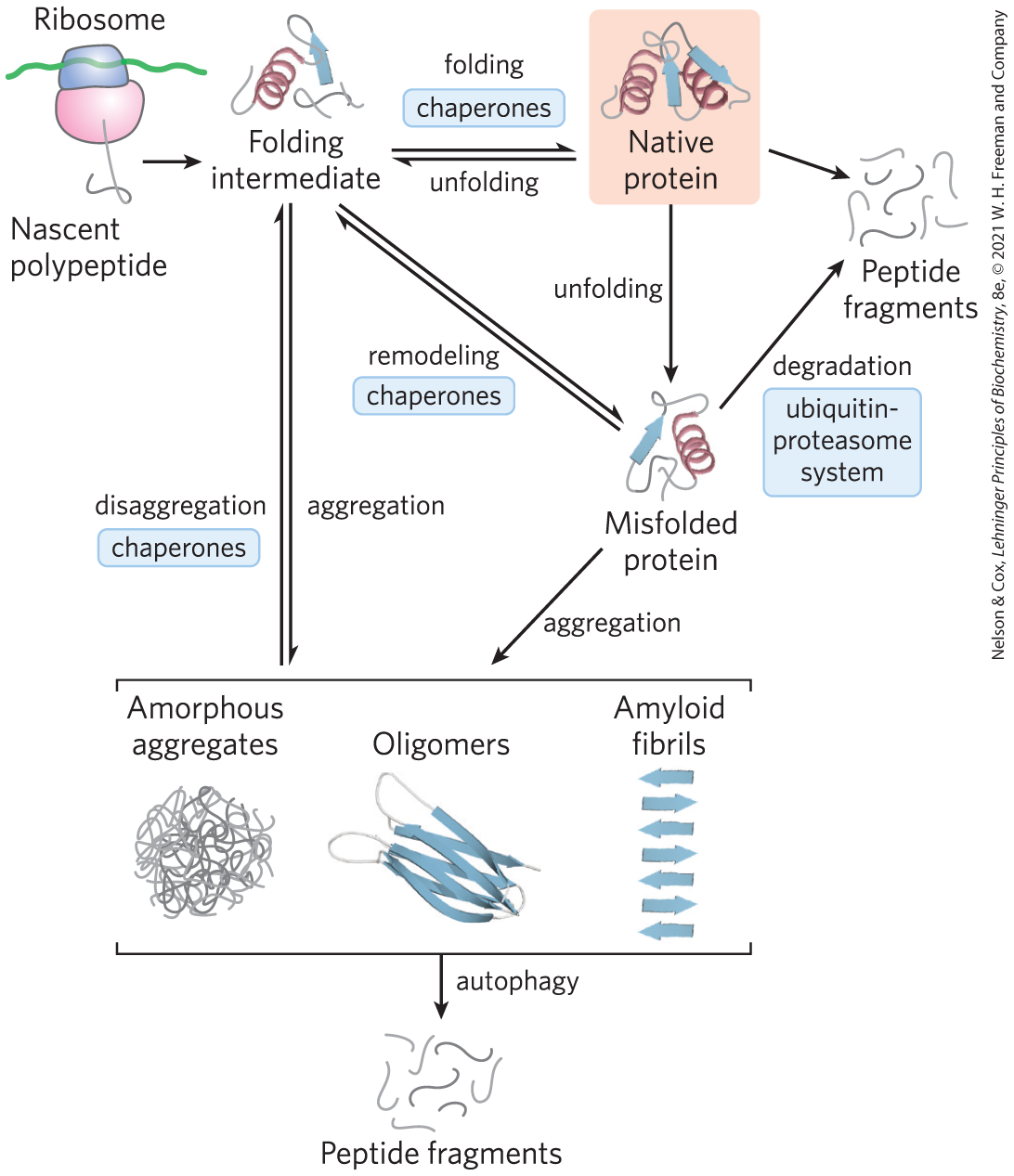
FIGURE 4-23 Pathways that contribute to proteostasis. Three kinds of processes contribute to proteostasis, in some cases with multiple contributing pathways. First, proteins are synthesized on a ribosome. Second, various pathways contribute to protein folding, many of which involve the activity of complexes called chaperones. Chaperones (including chaperonins) also contribute to the refolding of proteins that are partially and transiently unfolded. Finally, proteins that are irreversibly unfolded are subject to sequestration and degradation by several additional pathways. Partially unfolded proteins and protein-folding intermediates that escape the quality-control activities of the chaperones and degradative pathways may aggregate, forming both disordered aggregates and ordered amyloidlike aggregates that contribute to disease and aging processes. [Information from F. U. Hartl et al., Nature 475:324, 2011, Fig. 6.]
The transitions between the folded and unfolded states, and the network of pathways that control these transitions, now become our focus.
Loss of Protein Structure Results in Loss of Function
Protein structures have evolved to function in particular cellular or extracellular environments. Conditions different from these environments can result in protein structural changes, large and small. A loss of three-dimensional structure sufficient to cause loss of function is called denaturation. The denatured state does not necessarily equate with complete unfolding of the protein and randomization of conformation. Under most conditions, denatured proteins exist in a set of partially folded states.
Most proteins can be denatured by heat, which has complex effects on many weak interactions in a protein (primarily on the hydrogen bonds). If the temperature is increased slowly, a protein’s conformation generally remains intact until an abrupt loss of structure (and function) occurs over a narrow temperature range (Fig. 4-24). The abruptness of the change suggests that unfolding is a cooperative process: loss of structure in one part of the protein destabilizes other parts. The effects of heat on proteins can be mitigated by structure. The very heat-stable proteins of thermophilic bacteria and archaea have evolved to function at the temperature of hot springs The folded structures of these proteins are often similar to those of proteins in other organisms, but take some of the principles outlined here to extremes. They often feature high densities of charged residues on their surfaces, even tighter hydrophobic packing in their interiors, and folds rendered less flexible by networks of ion pairs. Each of these features makes these proteins less susceptible to unfolding at high temperatures.
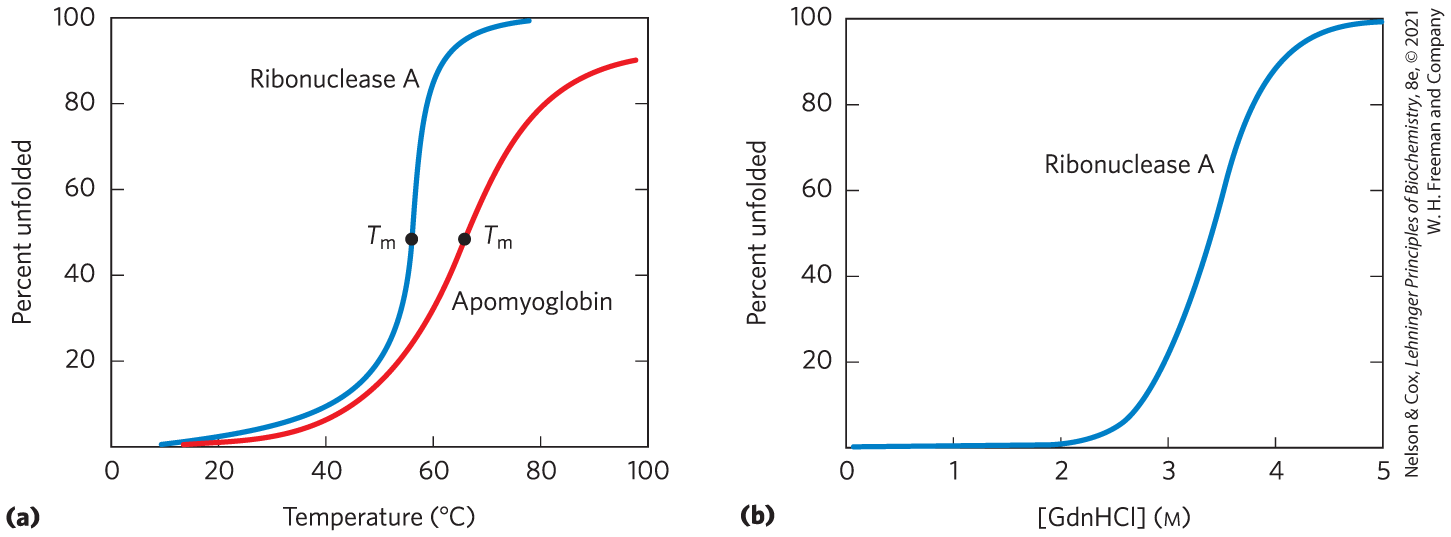
FIGURE 4-24 Protein denaturation. Results are shown for proteins denatured by two different environmental changes. In each case, the transition from the folded state to the unfolded state is abrupt, suggesting cooperativity in the unfolding process. (a) Thermal denaturation of horse apomyoglobin (myoglobin without the heme prosthetic group) and ribonuclease A (with its disulfide bonds intact; see Fig. 4-26). The midpoint of the temperature range over which denaturation occurs is called the melting temperature, or Denaturation of apomyoglobin was monitored by circular dichroism (see Fig. 4-9), which measures the amount of helical structure in the protein. Denaturation of ribonuclease A was tracked by monitoring changes in the intrinsic fluorescence of the protein, which is affected by changes in the environment of a Trp residue introduced by mutation. (b) Denaturation of disulfide-intact ribonuclease A by guanidine hydrochloride (GdnHCl), monitored by circular dichroism. [Data from (a) R. A. Sendak et al., Biochemistry 35:12,978, 1996; I. Nishii et al., J. Mol. Biol. 250:223, 1995; (b) W. A. Houry et al., Biochemistry 35:10,125, 1996.]
Proteins can also be denatured by extremes of pH, by certain miscible organic solvents such as alcohol or acetone, by certain solutes such as urea and guanidine hydrochloride, or by detergents. Each of these denaturing agents represents a relatively mild treatment in the sense that no covalent bonds in the polypeptide chain are broken. Organic solvents, urea, and detergents act primarily by disrupting the hydrophobic aggregation of nonpolar amino acid side chains that produces the stable core of globular proteins; urea also disrupts hydrogen bonds; and extremes of pH alter the net charge on a protein, causing electrostatic repulsion and the disruption of some hydrogen bonding. The denatured structures resulting from these various treatments are not necessarily the same.
Denaturation often leads to protein precipitation, a consequence of protein aggregate formation as exposed hydrophobic surfaces associate. The aggregates are often highly disordered. One example is the protein precipitate that can be seen after an egg white is boiled. More-ordered aggregates are also observed in some proteins, as we shall see.
Amino Acid Sequence Determines Tertiary Structure
The tertiary structure of a globular protein is determined by its amino acid sequence. The most important proof of this came from experiments showing that denaturation of some proteins is reversible. Certain globular proteins denatured by heat, extremes of pH, or denaturing reagents will regain their native structure and their biological activity if they are returned to conditions in which the native conformation is stable. This process is called renaturation.
A classic example is the denaturation and renaturation of ribonuclease A, demonstrated by Christian Anfinsen in the 1950s. Purified ribonuclease A denatures completely in a concentrated urea solution in the presence of a reducing agent. The reducing agent cleaves the four disulfide bonds to yield eight Cys residues, and the urea disrupts the stabilizing hydrophobic effect, thus freeing the entire polypeptide from its folded conformation. Denaturation of ribonuclease is accompanied by a complete loss of catalytic activity. When the urea and the reducing agent are removed, the denatured ribonuclease spontaneously refolds into its correct tertiary structure, with full restoration of its catalytic activity (Fig. 4-25). The refolding of ribonuclease is so accurate that the four intrachain disulfide bonds are re-formed in the same positions in the renatured molecule as in the native ribonuclease. Later, similar results were obtained using chemically synthesized, catalytically active ribonuclease A. This eliminated the possibility that some minor contaminant in Anfinsen’s purified ribonuclease preparation might have contributed to renaturation of the enzyme, and thus it dispelled any remaining doubt that this enzyme folds spontaneously.
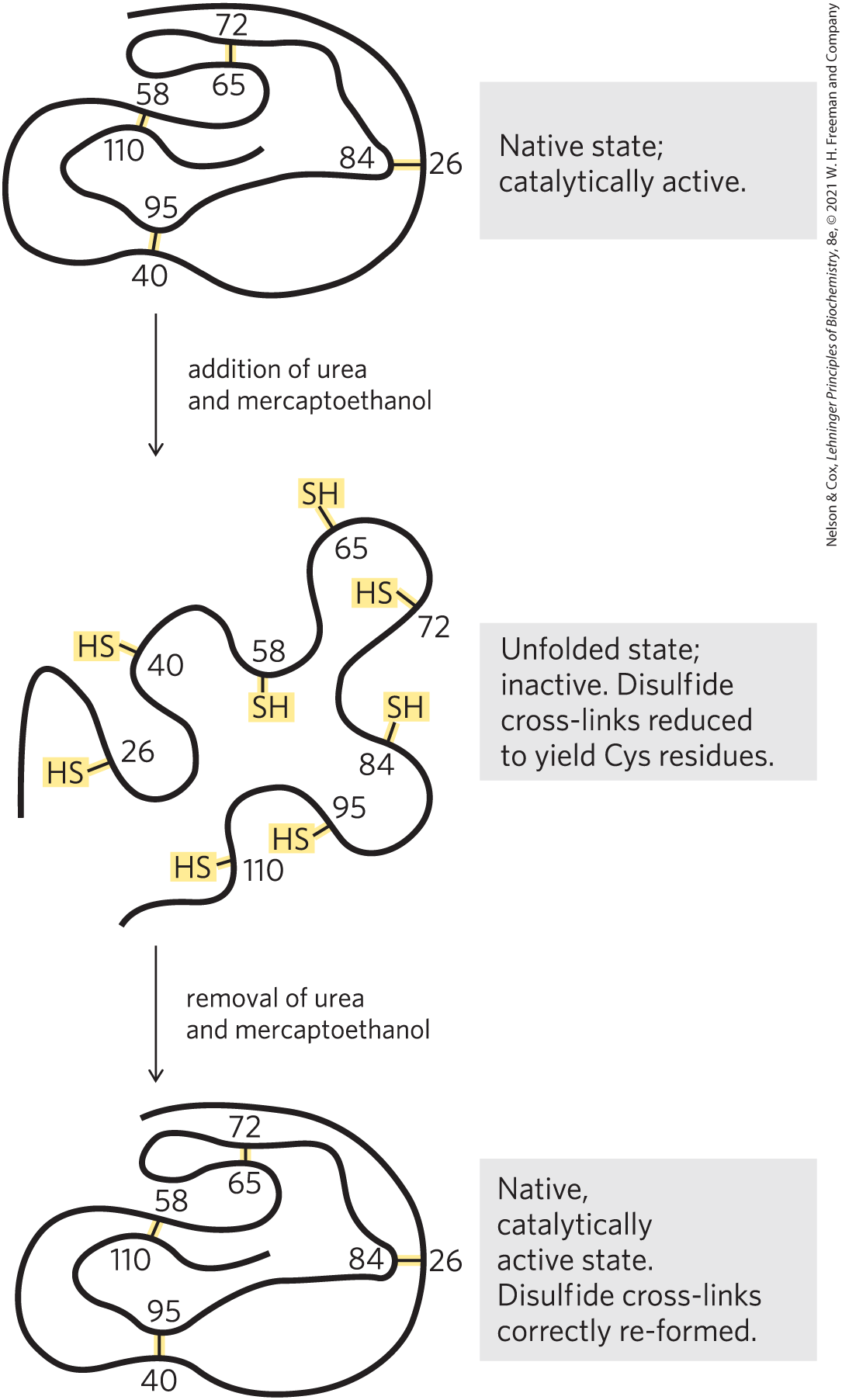
FIGURE 4-25 Renaturation of unfolded, denatured ribonuclease. Urea denatures the ribonuclease, and mercaptoethanol reduces and thus cleaves the disulfide bonds to yield eight Cys residues. Renaturation involves reestablishing the correct disulfide cross-links.
The Anfinsen experiment provided the first evidence that the amino acid sequence of a polypeptide chain contains all the information required to fold the chain into its native, three-dimensional structure. Subsequent work has shown that only a minority of proteins, many of them small and inherently stable, will fold spontaneously into their native form. Even though all proteins have the potential to fold into their native structure, many require some assistance.
Polypeptides Fold Rapidly by a Stepwise Process
In living cells, proteins are assembled from amino acids at a very high rate. For example, E. coli cells can make a complete, biologically active protein molecule containing 100 amino acid residues in about 5 seconds at However, the synthesis of peptide bonds on the ribosome is not enough; the protein must fold.
How does the polypeptide chain arrive at its native conformation? Let’s assume conservatively that each of the amino acid residues could take up 10 different conformations on average, giving different conformations for the polypeptide. Let’s also assume that the protein folds spontaneously by a random process in which it tries out all possible conformations around every single bond in its backbone until it finds its native, biologically active form. If each conformation were sampled in the shortest possible time ( second, or the time required for a single molecular vibration), it would take about years to sample all possible conformations! Clearly, protein folding is not a completely random, trial-and-error process. There must be shortcuts. This problem was first pointed out by Cyrus Levinthal in 1968 and is sometimes called Levinthal’s paradox.
The folding pathway of a large polypeptide chain is unquestionably complicated. However, robust algorithms can often predict the structure of smaller proteins on the basis of their amino acid sequences. The major folding pathways are hierarchical. Local secondary structures form first. Certain amino acid sequences fold readily into α helices or β sheets, guided by constraints such as those reviewed in our discussion of secondary structure. Ionic interactions, involving charged groups that are often near one another in the linear sequence of the polypeptide chain, can play an important role in guiding these early folding steps. Assembly of local structures is followed by longer-range interactions between, say, two elements of secondary structure that come together to form stable folded structures. The hydrophobic effect plays a significant role throughout the process, as the aggregation of nonpolar amino acid side chains provides an entropic stabilization to intermediates and, eventually, to the final folded structure. The process continues until complete domains form and the entire polypeptide is folded (Fig. 4-26). Notably, proteins dominated by close-range interactions (between pairs of residues generally located near each other in the polypeptide sequence) tend to fold faster than proteins with more complex folding patterns and with many long-range interactions between different segments. As larger proteins with multiple domains are synthesized, domains near the amino terminus (which are synthesized first) may fold before the entire polypeptide has been assembled.
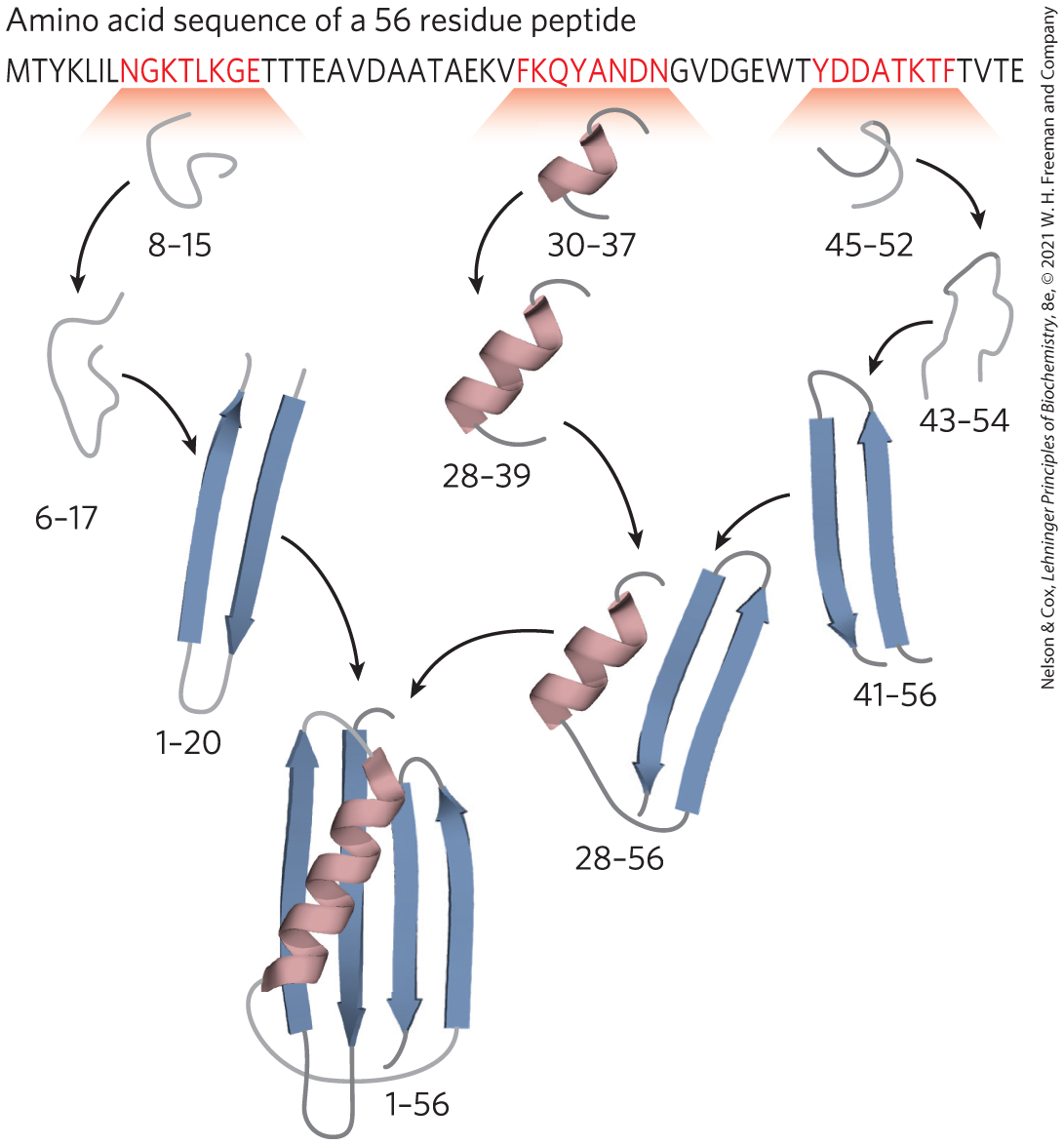
FIGURE 4-26 A protein-folding pathway as defined for a small protein. A hierarchical pathway is shown, based on computer modeling. Small regions of secondary structure are assembled first and then gradually incorporated into larger structures. The program used for this model has been highly successful in predicting the three-dimensional structure of small proteins from their amino acid sequence. The numbers indicate the amino acid residues in this 56 residue peptide that have acquired their final structure in each of the steps shown. [Information from K. A. Dill et al., Annu. Rev. Biophys. 37:289, 2008, Fig. 5.]
Thermodynamically, the folding process can be viewed as a kind of free-energy funnel (Fig. 4-27). The unfolded states are characterized by a high degree of conformational entropy and relatively high free energy. As folding proceeds, the narrowing of the funnel reflects the decrease in the conformational space that must be searched as the protein approaches its native state. Small depressions along the sides of the free-energy funnel represent semistable intermediates that can briefly slow the folding process. At the bottom of the funnel, an ensemble of folding intermediates has been reduced to a single native conformation (or one of a small set of native conformations). The funnels can have a variety of shapes, depending on the complexity of the folding pathway, the existence of semistable intermediates, and the potential for particular intermediates to assemble into aggregates of misfolded proteins.

FIGURE 4-27 The thermodynamics of protein folding depicted as free-energy funnels. As proteins fold, the conformational space that can be explored by the structure is constrained. This is modeled as a three-dimensional thermodynamic funnel, with ΔG represented by the depth of the funnel and the native structure (N) at the bottom (lowest free-energy point). The funnel for a given protein can have a variety of shapes, depending on the number and types of folding intermediates in the folding pathways. Any folding intermediate with significant stability and a finite lifetime would be represented as a local free-energy minimum—a depression on the surface of the funnel. (a) A simple but relatively wide and smooth funnel represents a protein that has multiple folding pathways (that is, the order in which different parts of the protein fold is somewhat random), but it assumes its three-dimensional structure with no folding intermediates that have significant stability. (b) This funnel represents a more typical protein that has multiple possible folding intermediates with significant stability on the multiple pathways leading to the native structure. [Information from K. A. Dill et al., Annu. Rev. Biophys. 37:289, 2008, Fig. 9.]
Thermodynamic stability is not evenly distributed over the structure of a protein — the molecule has regions of relatively high stability and others of low or negligible stability. For example, a protein may have two stable domains joined by a segment that is entirely disordered. Regions of low stability may allow a protein to alter its conformation between two or more states. As we shall see in the next two chapters, variations in the stability of regions within a protein are often essential to protein function. Intrinsically disordered proteins or protein segments do not fold at all.
Some Proteins Undergo Assisted Folding
Not all proteins fold spontaneously as they are synthesized in the cell. Folding for many proteins requires chaperones, proteins that interact with partially folded or improperly folded polypeptides, facilitating correct folding pathways or providing microenvironments in which folding can occur. Several types of molecular chaperones are found in organisms ranging from bacteria to humans. Two major families of chaperones, both well studied, are the Hsp70 family and the chaperonins.
Proteins in the Hsp70 family generally have a molecular weight near 70,000 and are more abundant in cells stressed by elevated temperatures (hence, heat shock proteins of 70,000, or Hsp70). Hsp70 proteins bind to regions of unfolded polypeptides that are rich in hydrophobic residues. These chaperones thus “protect” both proteins subject to denaturation by heat and new peptide molecules being synthesized (and not yet folded). Hsp70 proteins also block the folding of certain proteins that must remain unfolded until they have been translocated across a membrane (as described in Chapter 27). Some chaperones also facilitate the quaternary assembly of oligomeric proteins. The Hsp70 proteins bind to and release polypeptides in a cycle that uses energy from ATP hydrolysis and involves several other proteins (including a class called Hsp40). Figure 4-28 illustrates chaperone-assisted folding as elucidated for the eukaryotic Hsp70 and Hsp40 chaperones. The binding of an unfolded polypeptide by an Hsp70 chaperone may break up a protein aggregate or prevent the formation of a new one. When the bound polypeptide is released, it has a chance to resume folding to its native structure. If folding does not occur rapidly enough, the polypeptide may be bound again and the process repeated. Alternatively, the Hsp70-bound polypeptide may be delivered to a chaperonin.
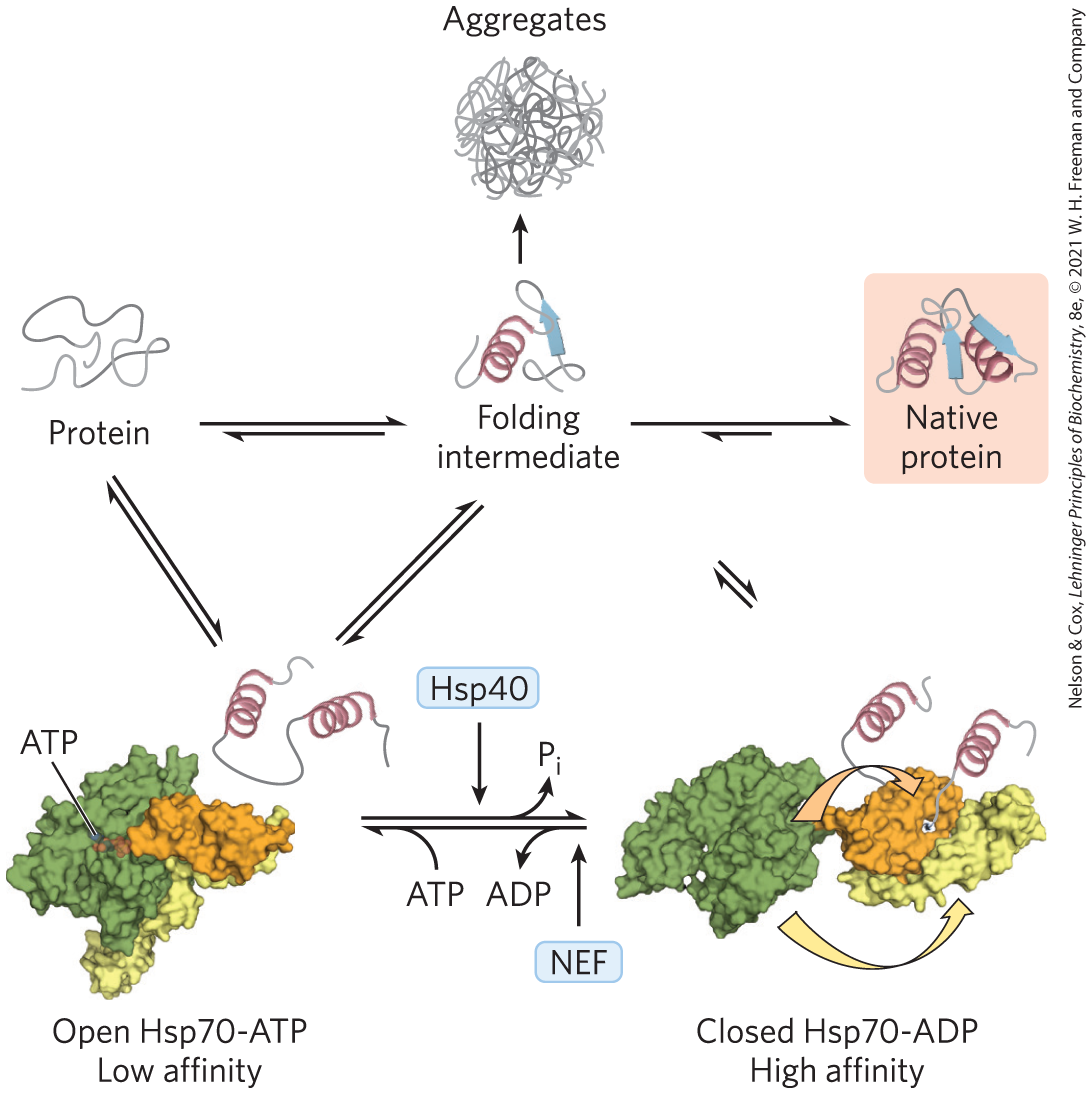
FIGURE 4-28 Chaperones in protein folding. The pathway by which chaperones of the Hsp70 class bind and release polypeptides is illustrated for the eukaryotic chaperones Hsp70 and Hsp40. The chaperones do not actively promote the folding of the substrate protein, but instead prevent aggregation of unfolded peptides. The unfolded or partly folded proteins bind first to the open, ATP-bound form of Hsp70. Hsp40 then interacts with this complex and triggers ATP hydrolysis that produces the closed form of the complex, in which the domains colored orange and yellow come together like the two parts of a jaw, trapping parts of the unfolded protein inside. Dissociation of ADP and recycling of the Hsp70 requires interaction with another type of protein called a nucleotide-exchange factor (NEF). For a population of polypeptide molecules, some fraction of the molecules released after the transient binding of partially folded proteins by Hsp70 will take up the native conformation. The remainder are quickly rebound by Hsp70 or diverted to the chaperonin system. [Information from F. U. Hartl et al., Nature 475:324, 2011, Fig. 2. Open Hsp70-ATP: PDB ID 2QXL, Q. Liu and W. A. Hendrickson, Cell 131:106, 2007. Closed Hsp70-ADP: Data from PDB ID 2KHO, E. B. Bertelson et al., Proc. Natl. Acad. Sci. USA 106:8471, 2009, and PDB ID 1DKZ, X. Zhu et al., Science 272:1606, 1996.]
Chaperonins are elaborate protein complexes required for the folding of some cellular proteins that do not fold spontaneously. In E. coli, an estimated 10% to 15% of cellular proteins require the resident chaperonin system, called GroEL/GroES, for folding under normal conditions (up to 30% require this assistance when the cells are heat stressed). The analogous chaperonin system in eukaryotes is called Hsp60. The chaperonins first became known when they were found to be necessary for the growth of certain bacterial viruses (hence the designation “Gro”). These chaperone proteins are structured as a series of multisubunit rings, forming two chambers oriented back to back. Inside one of the chambers, a protein is given about 10 seconds to fold. Constraining a protein within the chamber prevents inappropriate protein aggregation and also restricts the conformational space that a polypeptide chain can explore as it folds. The GroEL/GroES folding pathway is discussed in Chapter 27.
Finally, the folding pathways of some proteins require two enzymes that catalyze isomerization reactions. Protein disulfide isomerase (PDI) is a widely distributed enzyme that catalyzes the interchange, or shuffling, of disulfide bonds until the bonds of the native conformation are formed. Among its functions, PDI catalyzes the elimination of folding intermediates with inappropriate disulfide cross-links. Peptide prolyl cis-trans isomerase (PPI) catalyzes the interconversion of the cis and trans isomers of peptide bonds formed by Pro residues (Fig. 4-7), which can be a slow step in the folding of proteins that contain some Pro peptide bonds in the cis configuration.
Defects in Protein Folding Are the Molecular Basis for Many Human Genetic Disorders
Protein misfolding is a substantial problem in all cells. Despite the many processes that assist in protein folding, a quarter or more of all polypeptides synthesized may be destroyed because they do not fold correctly. In some cases, the misfolding causes or contributes to the development of serious disease.
 Protein misfolding is a substantial problem in all cells. Despite the many processes that assist in protein folding, a quarter or more of all polypeptides synthesized may be destroyed because they do not fold correctly.
Protein misfolding is a substantial problem in all cells. Despite the many processes that assist in protein folding, a quarter or more of all polypeptides synthesized may be destroyed because they do not fold correctly. Many conditions, including type 2 diabetes, Alzheimer disease, Huntington disease, and Parkinson disease, are associated with a misfolding mechanism: a soluble protein that is normally secreted from the cell is secreted in a misfolded state and converted into an insoluble extracellular amyloid fiber. The diseases are collectively referred to as amyloidoses. The fibers are highly ordered and unbranched, with a diameter of 7 to 10 nm and a high degree of β-sheet structure. The β segments are oriented perpendicular to the axis of the fiber. In some amyloid fibers the overall structure includes two layers of β sheet, such as that shown for amyloid-β peptide in Figure 4-29.
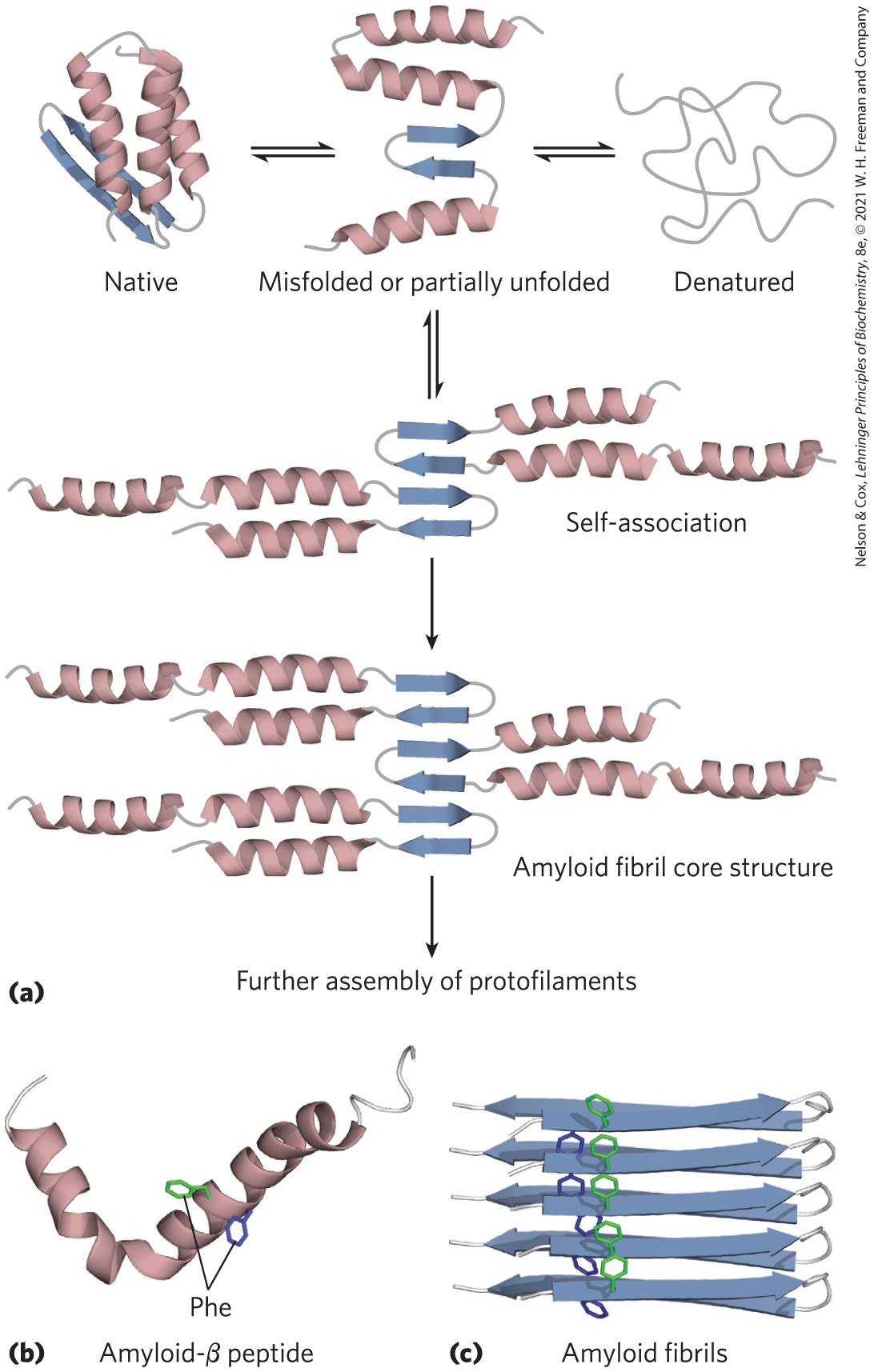
FIGURE 4-29 Formation of disease-causing amyloid fibrils. (a) Protein molecules whose normal structure includes regions of β sheet undergo partial folding. In a small number of the molecules, before folding is complete, the β-sheet regions of one polypeptide associate with the same region in another polypeptide, forming the nucleus of an amyloid. Additional protein molecules slowly associate with the amyloid and extend it to form a fibril. (b) The amyloid-β peptide begins as two α-helical segments of a larger protein. Proteolytic cleavage of this larger protein leaves the relatively unstable amyloid-β peptide, which loses its α-helical structure. It can then assemble slowly into amyloid fibrils (c), which contribute to the characteristic plaques on the exterior of nervous tissue in people with Alzheimer disease. The aromatic side chains shown here play a significant role in stabilizing the amyloid structure. Amyloid is rich in β sheet structure, with the β strands arranged perpendicular to the axis of the amyloid fibril. Amyloid-β peptide takes the form of two layers of extended parallel β sheet. [(a) Information from D. J. Selkoe, Nature 426:900, 2003, Fig. 1. (b) Data from PDB ID 1IYT, O. Crescenzi et al., Eur. J. Biochem. 269:5642, 2002. (c) Data from PDB ID 2BEG, T. Lührs et al., Proc. Natl. Acad. Sci. USA 102:17,342, 2005.]
Many proteins can take on the amyloid fibril structure as an alternative to their normal folded conformations, and most of these proteins have a concentration of aromatic amino acid residues in a core region of β sheet or α helix. The proteins are secreted in an incompletely folded conformation. The core (or some part of it) folds into a β sheet before the rest of the protein folds correctly, and the β sheets of two or more incompletely folded protein molecules associate to begin forming an amyloid fibril. The fibril grows in the extracellular space. Other parts of the protein then fold differently, remaining on the outside of the β-sheet core in the growing fibril. The effect of aromatic residues in stabilizing the structure is shown in Figure 4-29c. Because most of the protein molecules fold normally, the onset of symptoms in the amyloidoses is often very slow. If a person inherits a mutation such as substitution with an aromatic residue at a position that favors formation of amyloid fibrils, disease symptoms may begin at an earlier age.
The amyloid deposition diseases that trigger neurodegeneration, particularly in older adults, are a special class of localized amyloidoses. Alzheimer disease is associated with extracellular amyloid deposition by neurons, involving the amyloid-β peptide (Fig. 4-29b), derived from a larger transmembrane protein (amyloid-β precursor protein) found in most human tissues. When it is part of the larger protein, the peptide is composed of two α-helical segments spanning the membrane. When the external and internal domains are cleaved off by specific proteases, the relatively unstable amyloid-β peptide leaves the membrane and loses its α-helical structure. It can then take the form of two layers of extended parallel β sheet, which can slowly assemble into amyloid fibrils (Fig. 4-29c). Deposits of these amyloid fibers seem to be the primary cause of Alzheimer disease, but a second type of amyloidlike aggregation, involving a protein called tau, also occurs intracellularly (in neurons) in people with Alzheimer disease. Inherited mutations in the tau protein do not result in Alzheimer disease, but they cause a frontotemporal dementia and parkinsonism (a condition with symptoms resembling Parkinson disease) that can be equally devastating.
Several other neurodegenerative conditions involve intracellular aggregation of misfolded proteins. In Parkinson disease, the misfolded form of the protein α-synuclein aggregates into spherical filamentous masses called Lewy bodies. Huntington disease involves the protein huntingtin, which has a long polyglutamine repeat. In some individuals, the polyglutamine repeat is longer than normal, and a more subtle type of intracellular aggregation occurs. Notably, when the mutant human proteins involved in Parkinson disease and Huntington disease are expressed in Drosophila melanogaster, the flies display neurodegeneration expressed as eye deterioration, tremors, and early death. All of these symptoms are highly suppressed if expression of the Hsp70 chaperone is also increased.
Protein misfolding need not lead to amyloid formation to cause serious disease. For example, cystic fibrosis is caused by defects in a membrane-bound protein called cystic fibrosis transmembrane conductance regulator (CFTR), which acts as a channel for chloride ions. The most common cystic fibrosis–causing mutation is the deletion of a Phe residue at position 508 in CFTR, which causes improper protein folding. Most of this protein is then degraded and its normal function is lost (see Box 11-2). Drugs that can correct certain CFTR misfolding events have been developed. Such drugs are called “correctors” or “pharmacological chaperones.” Many of the disease-related mutations in collagen (p. 118) also cause defective folding. A particularly remarkable type of protein misfolding is seen in the prion diseases (Box 4-4).
SUMMARY 4.4 Protein Denaturation and Folding
- The maintenance of the steady-state collection of active cellular proteins required under a particular set of conditions — called proteostasis — involves an elaborate set of pathways and processes that fold, refold, and degrade polypeptide chains.
- The three-dimensional structure and the function of most proteins can be destroyed by denaturation, demonstrating a relationship between structure and function. Heat, extremes of pH, organic solvents, solutes, and detergents can all be used to denature proteins.
- Some denatured proteins can renature spontaneously to form biologically active protein, showing that tertiary structure is determined by amino acid sequence.
- Protein folding occurs too fast for it to be a completely random process. Instead, protein folding is generally hierarchical. Initially, regions of secondary structure may form, followed by folding into motifs and domains. Large ensembles of folding intermediates are rapidly brought to a single native conformation.
- For many proteins, folding is facilitated by Hsp70 chaperones and by chaperonins. Disulfide-bond formation and the cis-trans isomerization of Pro peptide bonds can also be catalyzed by specific enzymes during folding.
- Protein misfolding is the molecular basis for many human diseases, including cystic fibrosis and amyloidoses such as Alzheimer disease.
 The maintenance of the steady-state collection of active cellular proteins required under a particular set of conditions — called proteostasis — involves an elaborate set of pathways and processes that fold, refold, and degrade polypeptide chains.
The maintenance of the steady-state collection of active cellular proteins required under a particular set of conditions — called proteostasis — involves an elaborate set of pathways and processes that fold, refold, and degrade polypeptide chains.