Chapter 6 - Figures
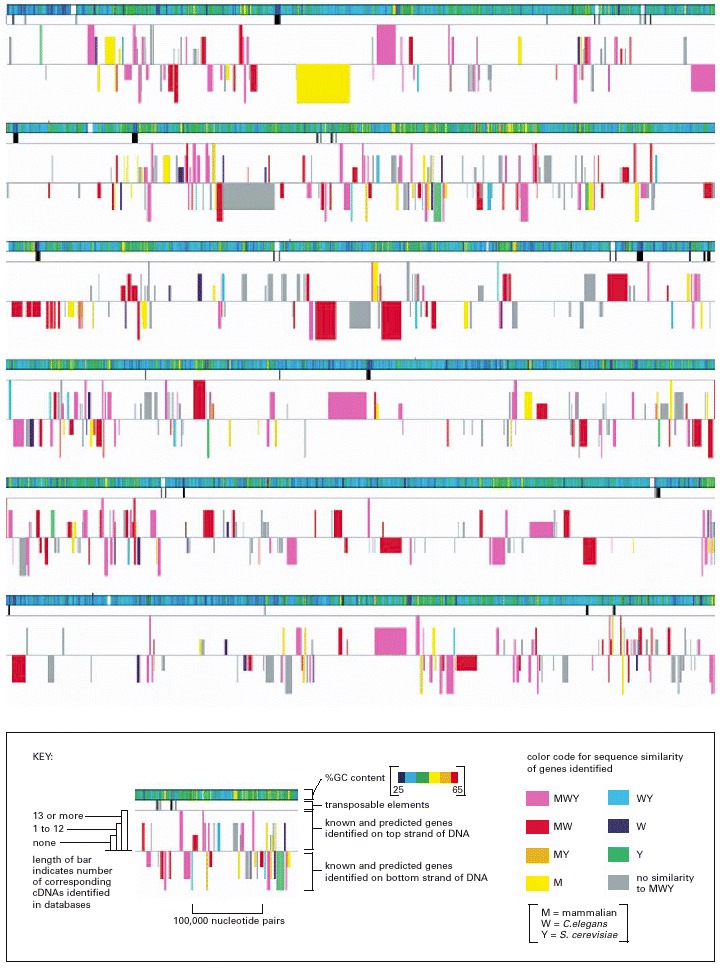
. This figure represents approximately 3% of the total Drosophila genome, arranged as six contiguous segments. As summarized in the key, the symbolic representations are: rainbow-colored bar: G-C base-pair content; black vertical lines of various thicknesses: locations of transposable elements, with thicker bars indicating clusters of elements; colored boxes: genes (both known and predicted) coded on one strand of DNA (boxes above the midline) and genes coded on the other strand (boxes below the midline). The length of each predicted gene includes both its exons (protein-coding DNA) and its introns (non-coding DNA) (see Figure 4-25). As indicated in the key, the height of each gene box is proportional to the number of cDNAs in various databases that match the gene. As described in Chapter 8, cDNAs are DNA copies of mRNA molecules, and large collections of the nucleotide sequences of cDNAs have been deposited in a variety of databases. The higher the number of matches between the nucleotide sequences of cDNAs and that of a particular predicted gene, the higher the confidence that the predicted gene is transcribed into RNA and is thus a genuine gene. The color of each gene box (see color code in the key) indicates whether a closely related gene is known to occur in other organisms. For example, MWY means the gene has close relatives in mammals, in the nematode worm Caenorhabditis elegans, and in the yeast Saccharomyces cerevisiae. MW indicates the gene has close relatives in mammals and the worm but not in yeast. (From Mark D. Adams et al., Science 287:2185–2195, 2000. © AAAS.)
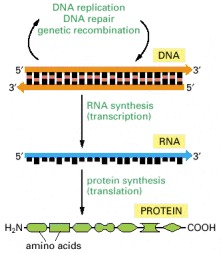
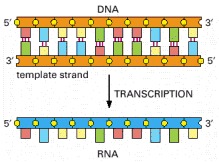
The flow of genetic information from DNA to RNA (transcription) and from RNA to protein (translation) occurs in all living cells.
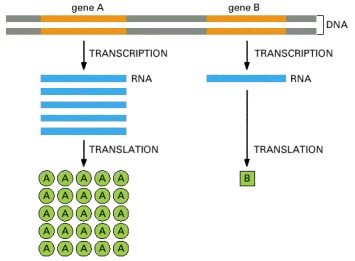
Gene A is transcribed and translated much more efficiently than gene B. This allows the amount of protein A in the cell to be much greater than that of protein B.
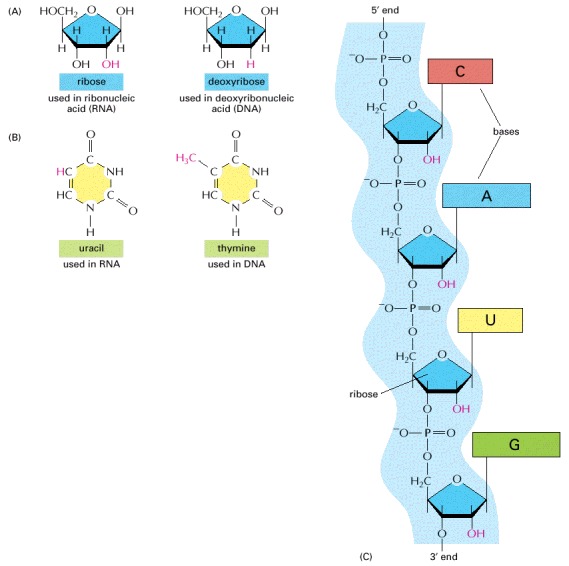
(A) RNA contains the sugar ribose, which differs from deoxyribose, the sugar used in DNA, by the presence of an additional -OH group. (B) RNA contains the base uracil, which differs from thymine, the equivalent base in DNA, by the absence of a -CH3 group. (C) A short length of RNA. The phosphodiester chemical linkage between nucleotides in RNA is the same as that in DNA.
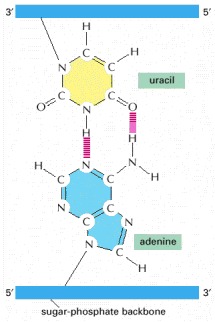
The absence of a methyl group in U has no effect on base-pairing; thus, U-A base pairs closely resemble T-A base pairs (see Figure 4-4).
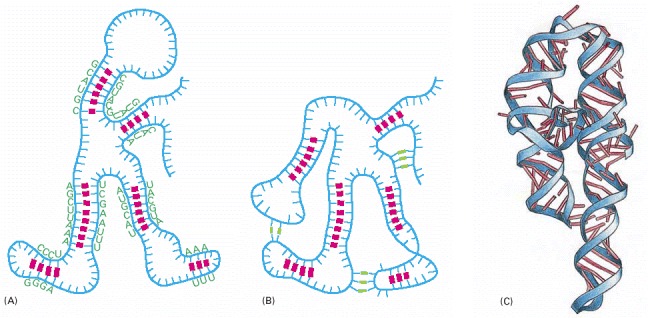
RNA is largely single-stranded, but it often contains short stretches of nucleotides that can form conventional base-pairs with complementary sequences found elsewhere on the same molecule. These interactions, along with additional “nonconventional” base-pair interactions, allow an RNA molecule to fold into a three-dimensional structure that is determined by its sequence of nucleotides. (A) Diagram of a folded RNA structure showing only conventional base-pair interactions; (B) structure with both conventional (red) and nonconventional (green) base-pair interactions; (C) structure of an actual RNA, a portion of a group 1 intron (see Figure 6-36). Each conventional base-pair interaction is indicated by a “rung” in the double helix. Bases in other configurations are indicated by broken rungs.
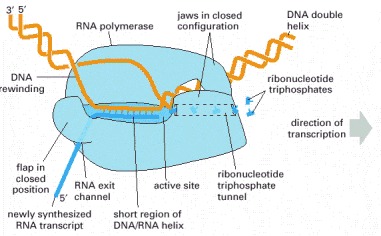
The RNA polymerase (pale blue) moves stepwise along the DNA, unwinding the DNA helix at its active site. As it progresses, the polymerase adds nucleotides (here, small “T” shapes) one by one to the RNA chain at the polymerization site using an exposed DNA strand as a template. The RNA transcript is thus a single-stranded complementary copy of one of the two DNA strands. The polymerase has a rudder (see Figure 6-11) that displaces the newly formed RNA, allowing the two strands of DNA behind the polymerase to rewind. A short region of DNA/RNA helix (approximately nine nucleotides in length) is therefore formed only transiently, and a “window” of DNA/RNA helix therefore moves along the DNA with the polymerase. The incoming nucleotides are in the form of ribonucleoside triphosphates (ATP, UTP, CTP, and GTP), and the energy stored in their phosphate-phosphate bonds provides the driving force for the polymerization reaction (see Figure 5-4). (Adapted from a figure kindly supplied by Robert Landick.)
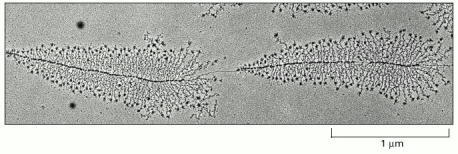
The micrograph shows many molecules of RNA polymerase simultaneously transcribing each of two adjacent genes. Molecules of RNA polymerase are visible as a series of dots along the DNA with the newly synthesized transcripts (fine threads) attached to them. The RNA molecules (ribosomal RNAs) shown in this example are not translated into protein but are instead used directly as components of ribosomes, the machines on which translation takes place. The particles at the 5′ end (the free end) of each rRNA transcript are believed to reflect the beginnings of ribosome assembly. From the lengths of the newly synthesized transcripts, it can be deduced that the RNA polymerase molecules are transcribing from left to right. (Courtesy of Ulrich Scheer.)
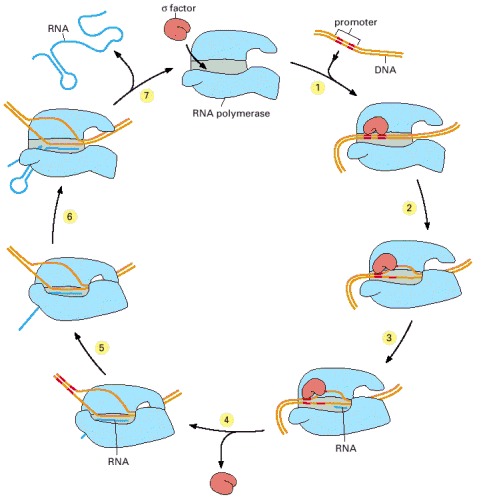
In step 1, the RNA polymerase holoenzyme (core polymerase plus σ factor) forms and then locates a promoter (see Figure 6-12). The polymerase unwinds the DNA at the position at which transcription is to begin (step 2) and begins transcribing (step 3). This initial RNA synthesis (sometimes called “abortive initiation”) is relatively inefficient. However, once RNA polymerase has managed to synthesize about 10 nucleotides of RNA, σ relaxes its grip, and the polymerase undergoes a series of conformational changes (which probably includes a tightening of its jaws and the placement of RNA in the exit channel [see Figure 6-11]). The polymerase now shifts to the elongation mode of RNA synthesis (step 4), moving rightwards along the DNA in this diagram. During the elongation mode (step 5) transcription is highly processive, with the polymerase leaving the DNA template and releasing the newly transcribed RNA only when it encounters a termination signal (step 6). Termination signals are encoded in DNA and many function by forming an RNA structure that destabilizes the polymerase's hold on the RNA, as shown here. In bacteria, all RNA molecules are synthesized by a single type of RNA polymerase and the cycle depicted in the figure therefore applies to the production of mRNAs as well as structural and catalytic RNAs. (Adapted from a figure kindly supplied by Robert Landick.)
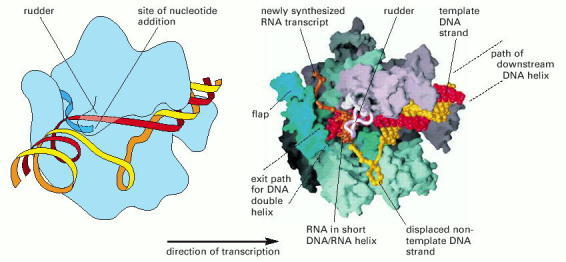
Two depictions of the three-dimensional structure of a bacterial RNA polymerase, with the DNA and RNA modeled in. This RNA polymerase is formed from four different subunits, indicated by different colors (right). The DNA strand used as a template is red, and the non-template strand is yellow. The rudder wedges apart the DNA-RNA hybrid as the polymerase moves. For simplicity only the polypeptide backbone of the rudder is shown in the right-hand figure, and the DNA exiting from the polymerase has been omitted. Because the RNA polymerase is depicted in the elongation mode, the σ factor is absent. (Courtesy of Seth Darst.)
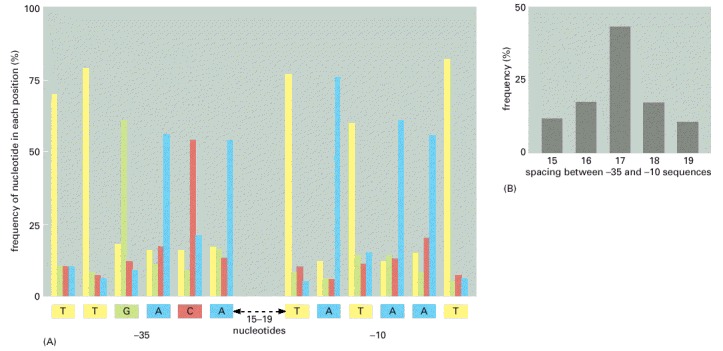
(A) The promoters are characterized by two hexameric DNA sequences, the -35 sequence and the -10 sequence named for their approximate location relative to the start point of transcription (designated +1). For convenience, the nucleotide sequence of a single strand of DNA is shown; in reality the RNA polymerase recognizes the promoter as double-stranded DNA. On the basis of a comparison of 300 promoters, the frequencies of the four nucleotides at each position in the -35 and -10 hexamers are given. The consensus sequence, shown below the graph, reflects the most common nucleotide found at each position in the collection of promoters. The sequence of nucleotides between the -35 and -10 hexamers shows no significant similarities among promoters. (B) The distribution of spacing between the -35 and -10 hexamers found in E. coli promoters. The information displayed in these two graphs applies to E. coli promoters that are recognized by RNA polymerase and the major σ factor (designated σ70). As we shall see in the next chapter, bacteria also contain minor σ factors, each of which recognizes a different promoter sequence. Some particularly strong promoters recognized by RNA polymerase and σ70 have an additional sequence, located upstream (to the left, in the figure) of the -35 hexamer, which is recognized by another subunit of RNA polymerase.
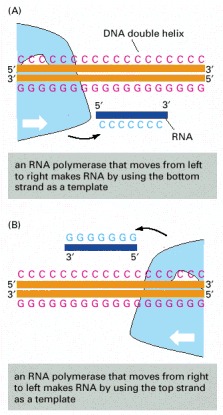
The DNA strand serving as template must be traversed in a 3′ to 5′ direction, as illustrated in Figure 6-9. Thus, the direction of RNA polymerase movement determines which of the two DNA strands is to serve as a template for the synthesis of RNA, as shown in (A) and (B). Polymerase direction is, in turn, determined by the orientation of the promoter sequence, the site at which the RNA polymerase begins transcription.
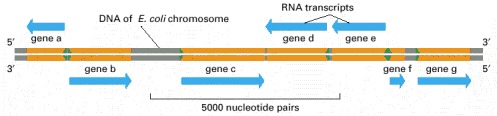
Some genes are transcribed using one DNA strand as a template, while others are transcribed using the other DNA strand. The direction of transcription is determined by the promoter at the beginning of each gene (green arrowheads). Approximately 0.2% (9000 base pairs) of the E. coli chromosome is depicted here. The genes transcribed from left to right use the bottom DNA strand as the template; those transcribed from right to left use the top strand as the template.
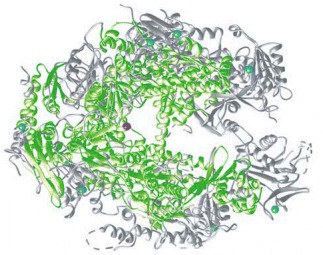
Regions of the two RNA polymerases that have similar structures are indicated in green. The eucaryotic polymerase is larger than the bacterial enzyme (12 subunits instead of 5), and some of the additional regions are shown in gray. The blue spheres represent Zn atoms that serve as structural components of the polymerases, and the red sphere represents the Mg atom present at the active site, where polymerization takes place. The RNA polymerases in all modern-day cells (bacteria, archaea, and eucaryotes) are closely related, indicating that the basic features of the enzyme were in place before the divergence of the three major branches of life. (Courtesy of P. Cramer and R. Kornberg.)
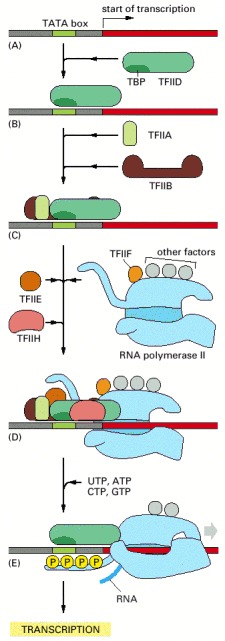
To begin transcription, RNA polymerase requires a number of general transcription factors (called TFIIA, TFIIB, and so on). (A) The promoter contains a DNA sequence called the TATA box, which is located 25 nucleotides away from the site at which transcription is initiated. (B) The TATA box is recognized and bound by transcription factor TFIID, which then enables the adjacent binding of TFIIB (C). For simplicity the DNA distortion produced by the binding of TFIID (see Figure 6-18) is not shown. (D) The rest of the general transcription factors, as well as the RNA polymerase itself, assemble at the promoter. (E) TFIIH then uses ATP to pry apart the DNA double helix at the transcription start point, allowing transcription to begin. TFIIH also phosphorylates RNA polymerase II, changing its conformation so that the polymerase is released from the general factors and can begin the elongation phase of transcription. As shown, the site of phosphorylation is a long C-terminal polypeptide tail that extends from the polymerase molecule. The assembly scheme shown in the figure was deduced from experiments performed in vitro, and the exact order in which the general transcription factors assemble on promoters in cells is not known with certainty. In some cases, the general factors are thought to first assemble with the polymerase, with the whole assembly subsequently binding to the DNA in a single step. The general transcription factors have been highly conserved in evolution; some of those from human cells can be replaced in biochemical experiments by the corresponding factors from simple yeasts.
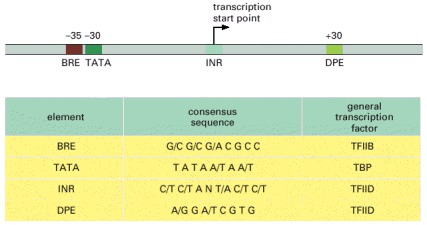
The name given to each consensus sequence (first column) and the general transcription factor that recognizes it (last column) are indicated. N indicates any nucleotide, and two nucleotides separated by a slash indicate an equal probability of either nucleotide at the indicated position. In reality, each consensus sequence is a shorthand representation of a histogram similar to that of Figure 6-12. For most RNA polymerase II transcription start points, only two or three of the four sequences are present. For example, most polymerase II promoters have a TATA box sequence, and those that do not typically have a “strong” INR sequence. Although most of the DNA sequences that influence transcription initiation are located “upstream” of the transcription start point, a few, such as the DPE shown in the figure, are located in the transcribed region.
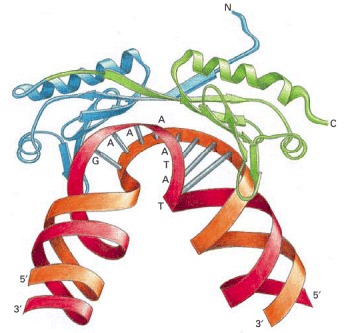
The TBP is the subunit of the general transcription factor TFIID that is responsible for recognizing and binding to the TATA box sequence in the DNA (red). The unique DNA bending caused by TBP—two kinks in the double helix separated by partly unwound DNA—may serve as a landmark that helps to attract the other general transcription factors. TBP is a single polypeptide chain that is folded into two very similar domains (blue and green). (Adapted from J.L. Kim et al., Nature 365:520–527, 1993.)
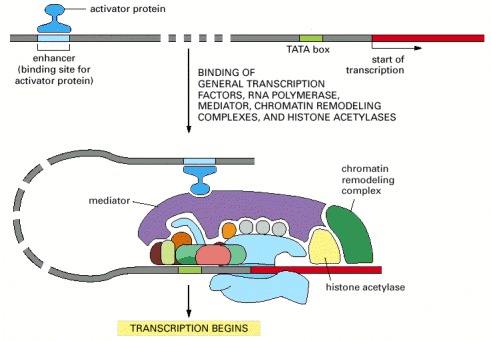
Transcription initiation in vivo requires the presence of transcriptional activator proteins. As described in Chapter 7, these proteins bind to specific short sequences in DNA. Although only one is shown here, a typical eucaryotic gene has many activator proteins, which together determine its rate and pattern of transcription. Sometimes acting from a distance of several thousand nucleotide pairs (indicated by the dashed DNA molecule), these gene regulatory proteins help RNA polymerase, the general factors, and the mediator all to assemble at the promoter. In addition, activators attract ATP-dependent chromatin-remodeling complexes and histone acetylases.
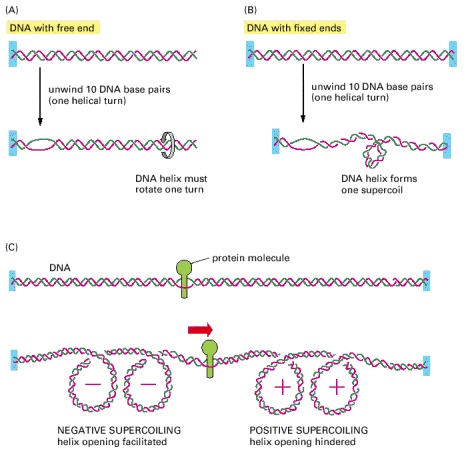
(A) For a DNA molecule with one free end (or a nick in one strand that serves as a swivel), the DNA double helix rotates by one turn for every 10 nucleotide pairs opened. (B) If rotation is prevented, superhelical tension is introduced into the DNA by helix opening. One way of accommodating this tension would be to increase the helical twist from 10 to 11 nucleotide pairs per turn in the double helix that remains in this example; the DNA helix, however, resists such a deformation in a springlike fashion, preferring to relieve the superhelical tension by bending into supercoiled loops. As a result, one DNA supercoil forms in the DNA double helix for every 10 nucleotide pairs opened. The supercoil formed in this case is a positive supercoil. (C) Supercoiling of DNA is induced by a protein tracking through the DNA double helix. The two ends of the DNA shown here are unable to rotate freely relative to each other, and the protein molecule is assumed also to be prevented from rotating freely as it moves. Under these conditions, the movement of the protein causes an excess of helical turns to accumulate in the DNA helix ahead of the protein and a deficit of helical turns to arise in the DNA behind the protein, as shown.
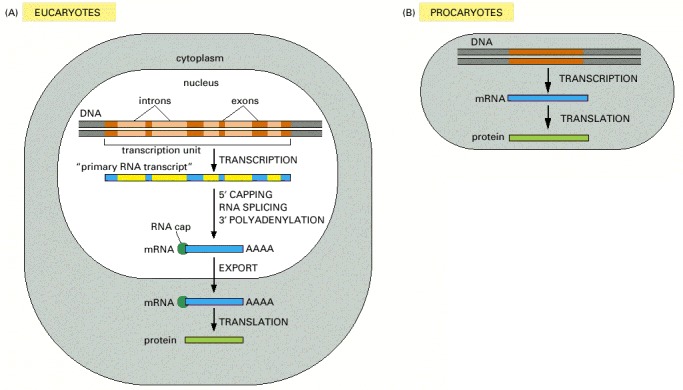
The final level of a protein in the cell depends on the efficiency of each step and on the rates of degradation of the RNA and protein molecules. (A) In eucaryotic cells the RNA molecule produced by transcription alone (sometimes referred to as the primary transcript) would contain both coding (exon) and noncoding (intron) sequences. Before it can be translated into protein, the two ends of the RNA are modified, the introns are removed by an enzymatically catalyzed RNA splicing reaction, and the resulting mRNA is transported from the nucleus to the cytoplasm. Although these steps are depicted as occurring one at a time, in a sequence, in reality they are coupled and different steps can occur simultaneously. For example, the RNA cap is added and splicing typically begins before transcription has been completed. Because of this coupling, complete primary RNA transcripts do not typically exist in the cell. (B) In procaryotes the production of mRNA molecules is much simpler. The 5′ end of an mRNA molecule is produced by the initiation of transcription by RNA polymerase, and the 3′ end is produced by the termination of transcription. Since procaryotic cells lack a nucleus, transcription and translation take place in a common compartment. In fact, translation of a bacterial mRNA often begins before its synthesis has been completed.
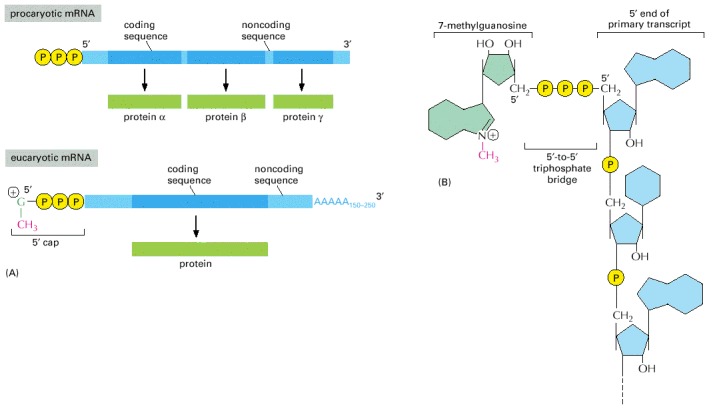
(A) The 5′ and 3′ ends of a bacterial mRNA are the unmodified ends of the chain synthesized by the RNA polymerase, which initiates and terminates transcription at those points, respectively. The corresponding ends of a eucaryotic mRNA are formed by adding a 5′ cap and by cleavage of the pre-mRNA transcript and the addition of a poly-A tail, respectively. The figure also illustrates another difference between the procaryotic and eucaryotic mRNAs: bacterial mRNAs can contain the instructions for several different proteins, whereas eucaryotic mRNAs nearly always contain the information for only a single protein. (B) The structure of the cap at the 5′ end of eucaryotic mRNA molecules. Note the unusual 5′-to-5′ linkage of the 7-methyl G to the remainder of the RNA. Many eucaryotic mRNAs carry an additional modification: the 2′-hydroxyl group on the second ribose sugar in the mRNA is methylated (not shown).
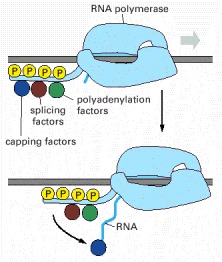
Not only does the polymerase transcribe DNA into RNA, but it also carries pre-mRNA-processing proteins on its tail, which are then transferred to the nascent RNA at the appropriate time. There are many RNA-processing enzymes, and not all travel with the polymerase. For RNA splicing, for example, only a few critical components are carried on the tail; once transferred to an RNA molecule, they serve as a nucleation site for the remaining components. The RNA-processing proteins first bind to the RNA polymerase tail when it is phosphorylated late in the process of transcription initiation (see Figure 6-16). Once RNA polymerase II finishes transcribing, it is released from DNA, the phosphates on its tail are removed by soluble phosphatases, and it can reinitiate transcription. Only this dephosphorylated form of RNA polymerase II is competent to start RNA synthesis at a promoter.
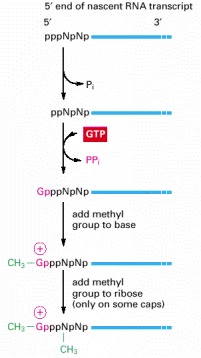
The final cap contains a novel 5′-to-5′ linkage between the positively charged 7-methyl G residue and the 5′ end of the RNA transcript (see Figure 6-22B). The letter N represents any one of the four ribonucleotides, although the nucleotide that starts an RNA chain is usually a purine (an A or a G). (After A.J. Shatkin, BioEssays 7:275–277, 1987. © ICSU Press.)
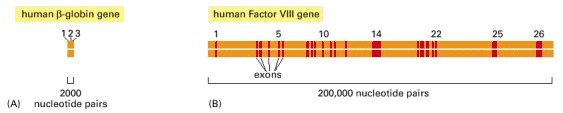
(A) The relatively small β-globin gene, which encodes one of the subunits of the oxygen-carrying protein hemoglobin, contains 3 exons (see also Figure 4-7). (B) The much larger Factor VIII gene contains 26 exons; it codes for a protein (Factor VIII) that functions in the blood-clotting pathway. Mutations in this gene are responsible for the most prevalent form of hemophilia.
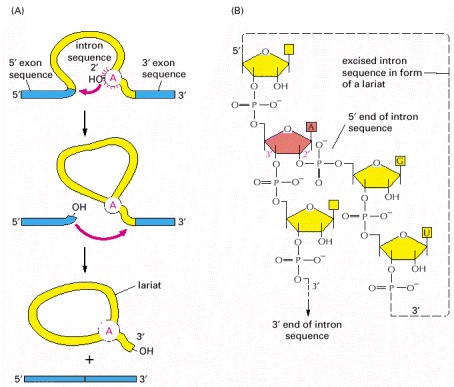
(A) In the first step, a specific adenine nucleotide in the intron sequence (indicated in red) attacks the 5′ splice site and cuts the sugar-phosphate backbone of the RNA at this point. The cut 5′ end of the intron becomes covalently linked to the adenine nucleotide, as shown in detail in (B), thereby creating a loop in the RNA molecule. The released free 3′-OH end of the exon sequence then reacts with the start of the next exon sequence, joining the two exons together and releasing the intron sequence in the shape of a lariat. The two exon sequences thereby become joined into a continuous coding sequence; the released intron sequence is degraded in due course.
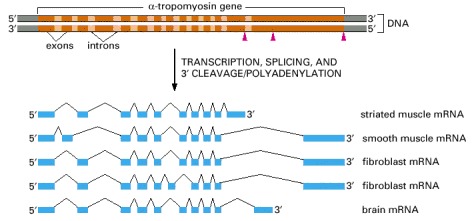
α-tropomyosin is a coiled-coil protein (see Figure 3-11) that regulates contraction in muscle cells. The primary transcript can be spliced in different ways, as indicated in the figure, to produce distinct mRNAs, which then give rise to variant proteins. Some of the splicing patterns are specific for certain types of cells. For example, the α-tropomyosin made in striated muscle is different from that made from the same gene in smooth muscle. The arrowheads in the top part of the figure demark the sites where cleavage and poly-A addition can occur.
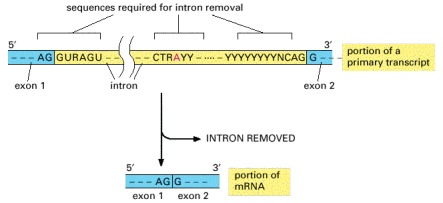
Only the three blocks of nucleotide sequences shown are required to remove an intron sequence; the rest of the intron can be occupied by any nucleotide. Here A, G, U, and C are the standard RNA nucleotides; R stands for either A or G; Y stands for either C or U. The A highlighted in red forms the branch point of the lariat produced by splicing. Only the GU at the start of the intron and the AG at its end are invariant nucleotides in the splicing consensus sequences. The remaining positions (even the branch point A) can be occupied by a variety of nucleotides, although the indicated nucleotides are preferred. The distances along the RNA between the three splicing consensus sequences are highly variable; however, the distance between the branch point and 3′ splice junction is typically much shorter than that between the 5′ splice junction and the branch point.
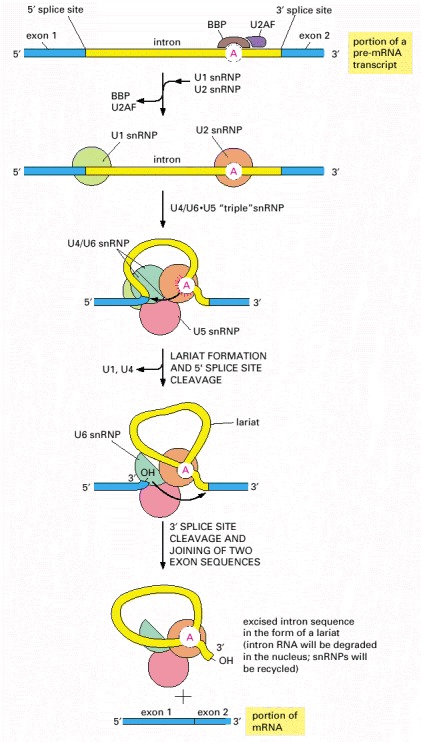
RNA splicing is catalyzed by an assembly of snRNPs (shown as colored circles) plus other proteins (most of which are not shown), which together constitute the spliceosome. The spliceosome recognizes the splicing signals on a pre-mRNA molecule, brings the two ends of the intron together, and provides the enzymatic activity for the two reaction steps (see Figure 6-26). The branch-point site is first recognized by the BBP (branch-point binding protein) and U2AF, a helper protein. In the next steps, the U2 snRNP displaces BBP and U2AF and forms base pairs with the branch-point site consensus sequence, and the U1 snRNP forms base-pairs with the 5′ splice junction (see Figure 6-30). At this point, the U4/U6•U5 “triple” snRNP enters the spliceosome. In this triple snRNP, the U4 and U6 snRNAs are held firmly together by base-pair interactions and the U5 snRNP is more loosely associated. Several RNA-RNA rearrangements then occur that break apart the U4/U6 base pairs (as shown, the U4 snRNP is ejected from the splicesome before splicing is complete) and allow the U6 snRNP to displace U1 at the 5′ splice junction (see Figure 6-30). Subsequent rearrangements create the active site of the spliceosome and position the appropriate portions of the pre-mRNA substrate for the splicing reaction to occur. Although not shown in the figure, each splicing event requires additional proteins, some of which hydrolyze ATP and promote the RNA-RNA rearrangements.
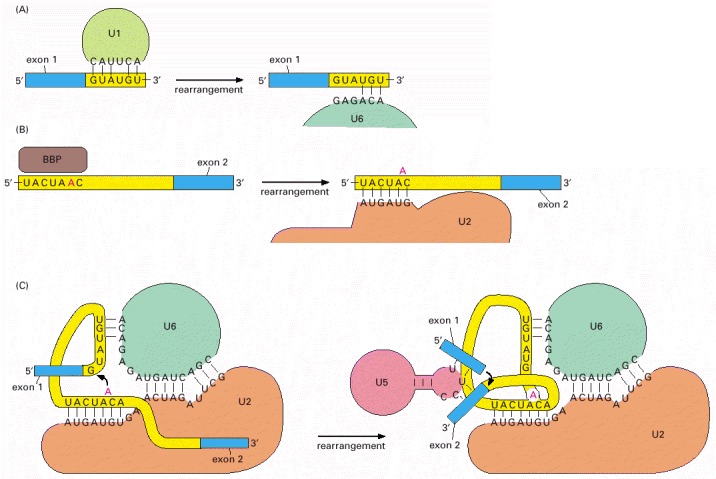
Shown here are the details for the yeast Saccharomyces cerevisiae, in which the nucleotide sequences involved are slightly different from those in human cells. (A) The exchange of U1 snRNP for U6 snRNP occurs before the first phosphoryl-transfer reaction (see Figure 6-29). This exchange allows the 5′ splice site to be read by two different snRNPs, thereby increasing the accuracy of 5′ splice site selection by the spliceosome. (B) The branch-point site is first recognized by BBP and subsequently by U2 snRNP; as in (A), this “check and recheck” strategy provides increased accuracy of site selection. The binding of U2 to the branch-point forces the appropriate adenine (in red) to be unpaired and thereby activates it for the attack on the 5′ splice site (see Figure 6-29). This, in combination with recognition by BBP, is the way in which the spliceosome accurately chooses the adenine that is ultimately to form the branch point. (C) After the first phosphoryl-transfer reaction (left) has occurred, U5 snRNP undergoes a rearrangement that brings the two exons into close proximity for the second phosphoryl-transfer reaction (right). The snRNAs both position the reactants and provide (either all or in part) the catalytic site for the two reactions. The U5 snRNP is present in the spliceosome before this rearrangement occurs; for clarity it has been omitted from the left panel. As discussed in the text, all of the RNA-RNA rearrangements shown in this figure (as well as others that occur in the spliceosome but are not shown) require the participation of additional proteins and ATP hydrolysis.
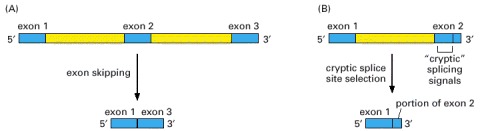
Both types might be expected to occur frequently if splice-site selection were performed by the spliceosome on a preformed, protein-free RNA molecule. “Cryptic” splicing signals are nucleotide sequences of RNA that closely resemble true splicing signals.
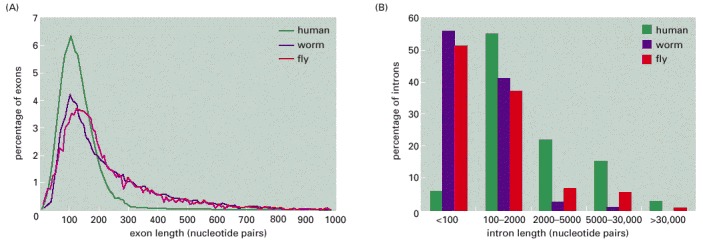
(A) Size distribution of exons. (B) Size distribution of introns. Note that exon length is much more uniform than intron length. (Adapted from International Human Genome Sequencing Consortium, Nature 409:860–921, 2001.)
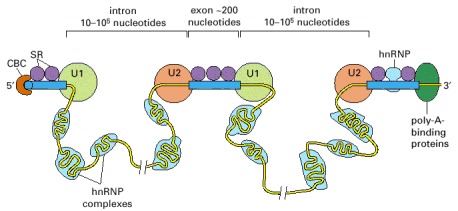
According to one proposal, SR proteins bind to each exon sequence in the pre-mRNA and thereby help to guide the snRNPs to the proper intron/exon boundaries. This demarcation of exons by the SR proteins occurs co-transcriptionally, beginning at the CBC (cap-binding complex) at the 5′ end. As indicated, the intron sequences in the pre-mRNA, which can be extremely long, are packaged into hnRNP (heterogeneous nuclear ribonucleoprotein) complexes that compact them into more manageable structures and perhaps mask cryptic splice sites. Each hnRNP complex forms a particle approximately twice the diameter of a nucleosome, and the core is composed of a set of at least eight different proteins. It has been proposed that hnRNP proteins preferentially associate with intron sequences and that this preference also helps the spliceosome distinguish introns from exons. However, as shown, at least some hnRNP proteins may bind to exon sequences but their role, if any, in exon definition has yet to be established. (Adapted from R. Reed, Curr. Opin. Cell Biol. 12:340–345, 2000.)
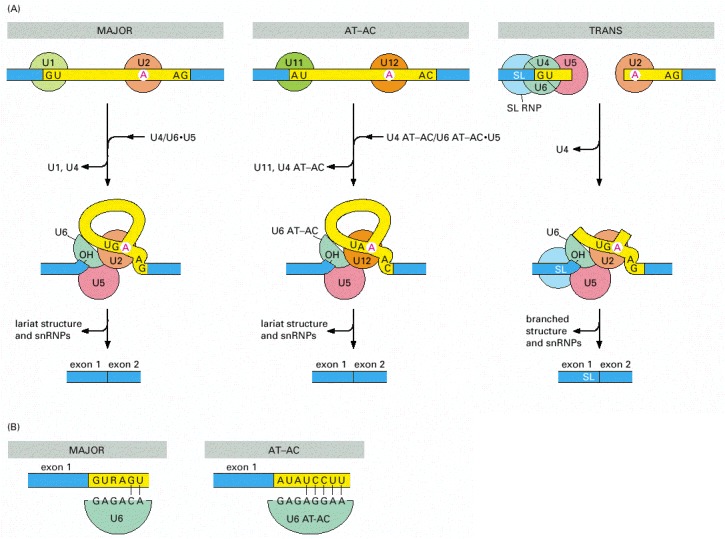
(A) Three types of spliceosomes. The major spliceosome (left), the AT-AC spliceosome (middle), and the trans-spliceosome (right) are each shown at two stages of assembly. The U5 snRNP is the only component that is common to all three spliceosomes. Introns removed by the AT-AC spliceosome have a different set of consensus nucleotide sequences from those removed by the major spliceosome. In humans, it is estimated that 0.1% of introns are removed by the AT-AC spliceosome. In trans-splicing, the SL snRNP is consumed in the reaction because a portion of the SL snRNA becomes the first exon of the mature mRNA. (B) The major U6 snRNP and the U6 AT-AC snRNP both recognize the 5′ splice junction, but they do so through a different set of base-pair interactions. The sequences shown are from humans. (Adapted from Y.-T. Yu et al., The RNA World, pp. 487–524. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, 1999.)
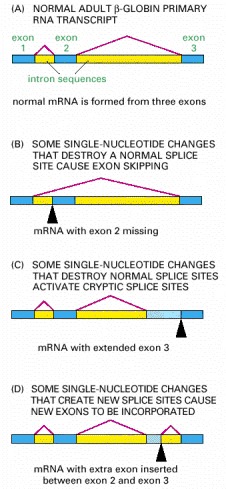
In the examples shown, the disease is caused by splice-site mutations, denoted by black arrowheads. The dark blue boxes represent the three normal exon sequences; the red lines are used to indicate the 5′ and 3′ splice sites that are used in splicing the RNA transcript. The light blue boxes depict new nucleotide sequences included in the final mRNA molecule as a result of the mutation. Note that when a mutation leaves a normal splice site without a partner, an exon is skipped or one or more abnormal “cryptic” splice sites nearby is used as the partner site, as in (C) and (D). (Adapted in part from S.H. Orkin, in The Molecular Basis of Blood Diseases [G. Stamatoyannopoulos et al., eds.], pp. 106–126. Philadelphia: Saunders, 1987.)
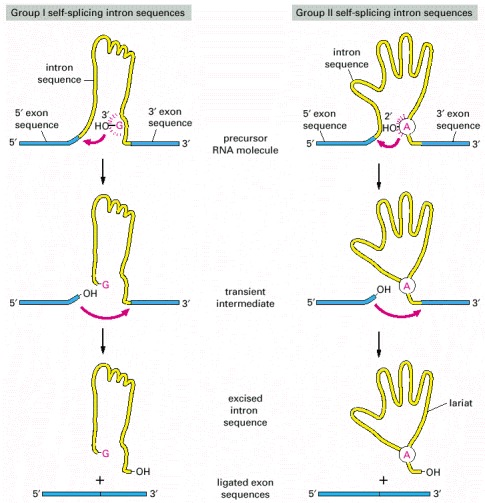
The group I intron sequences bind a free G nucleotide to a specific site on the RNA to initiate splicing, while the group II intron sequences use an especially reactive A nucleotide in the intron sequence itself for the same purpose. The two mechanisms have been drawn to emphasize their similarities. Both are normally aided in the cell by proteins that speed up the reaction, but the catalysis is nevertheless mediated by the RNA in the intron sequence. Both types of self-splicing reactions require the intron to be folded into a highly specific three-dimensional structure that provides the catalytic activity for the reaction (see Figure 6-6). The mechanism used by group II intron sequences releases the intron as a lariat structure and closely resembles the pathway of pre-mRNA splicing catalyzed by the spliceosome (compare with Figure 6-29). The great majority of RNA splicing in eucaryotic cells is performed by the spliceosome, and self-splicing RNAs represent unusual cases. (Adapted from T.R. Cech, Cell 44:207–210, 1986.)
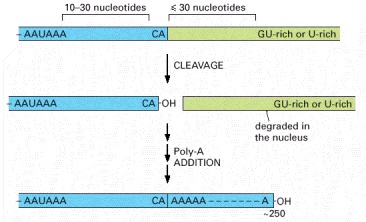
These sequences are encoded in the genome and are recognized by specific proteins after they are transcribed into RNA. The hexamer AAUAAA is bound by CPSF, the GU-rich element beyond the cleavage site by CstF (see Figure 6-38), and the CA sequence by a third factor required for the cleavage step. Like other consensus nucleotide sequences discussed in this chapter (see Figure 6-12), the sequences shown in the figure represent a variety of individual cleavage and polyadenylation signals.
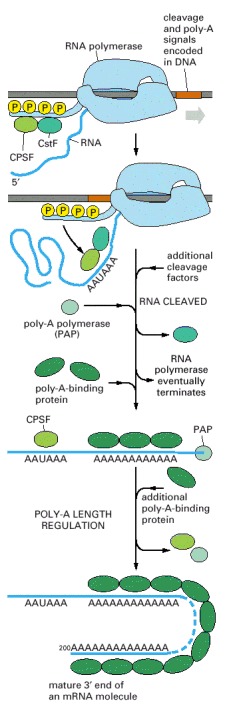
This process is much more complicated than the analogous process in bacteria, where the RNA polymerase simply stops at a termination signal and releases both the 3′ end of its transcript and the DNA template (see Figure 6-10).
(A) The maturation of a Balbiani Ring mRNA molecule as it is synthesized by RNA polymerase and packaged by a variety of nuclear proteins. This drawing of unusually abundant RNA produced by an insect cell is based on EM micrographs such as that shown in (B). Balbiani Rings are described in Chapter 4. (A, adapted from B. Daneholt, Cell 88:585–588, 1997; B, from B.J. Stevens and H. Swift, J. Cell Biol. 31:55–77, 1966. © The Rockefeller University Press.)
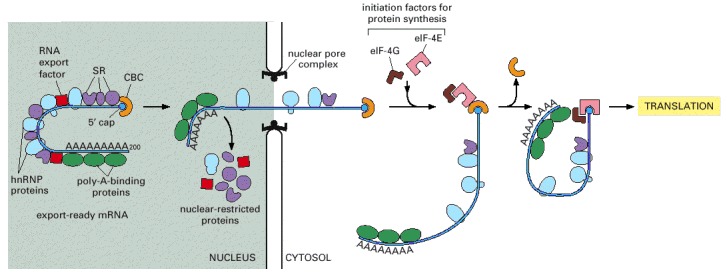
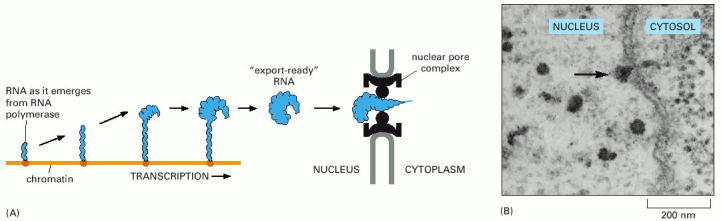
As indicated, some proteins travel with the mRNA as it moves through the pore, whereas others remain in the nucleus. Once in the cytoplasm, the mRNA continues to shed previously bound proteins and acquire new ones; these substitutions affect the subsequent translation of the message. Because some are transported with the RNA, the proteins that become bound to an mRNA in the nucleus can influence its subsequent stability and translation in the cytosol. RNA export factors, shown in the nucleus, play an active role in transporting the mRNA to the cytosol (see Figure 12-16). Some are deposited at exon-exon boundaries as splicing is completed, thus signifying those regions of the RNA that have been properly spliced.
The pattern of alternating transcribed gene and nontranscribed spacer is readily seen. A higher-magnification view was shown in Figure 6-9. (From V.E. Foe, Cold Spring Harbor Symp. Quant. Biol. 42:723–740, 1978.)
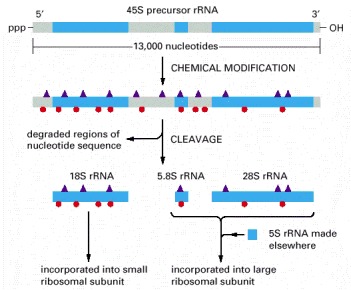
As indicated, two types of chemical modifications (shown in Figure 6-43) are made to the precursor rRNA before it is cleaved. Nearly half of the nucleotide sequences in this precursor rRNA are discarded and degraded in the nucleus. The rRNAs are named according to their “S” values, which refer to their rate of sedimentation in an ultra-centrifuge. The larger the S value, the larger the rRNA.
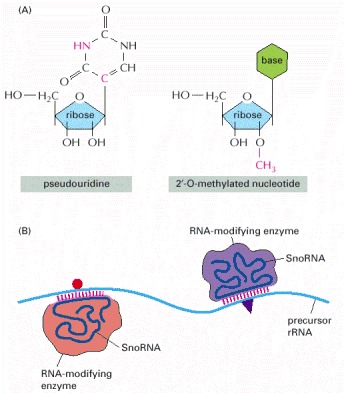
(A) Two prominent covalent modifications occur after rRNA synthesis; the differences from the initially incorporated nucleotide are indicated by red atoms. (B) As indicated, snoRNAs locate the sites of modification by base-pairing to complementary sequences on the precursor rRNA. The snoRNAs are bound to proteins, and the complexes are called snoRNPs. snoRNPs contain the RNA modification activities, presumably contributed by the proteins but possibly by the snoRNAs themselves.
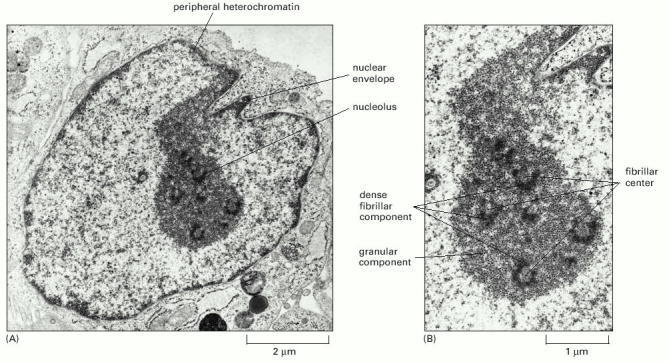
(A) View of entire nucleus. (B) High-power view of the nucleolus. It is believed that transcription of the rRNA genes takes place between the fibrillar center and the dense fibrillar component and that processing of the rRNAs and their assembly into ribosomes proceeds outward from the dense fibrillar component to the surrounding granular components. (Courtesy of E.G. Jordan and J. McGovern.)
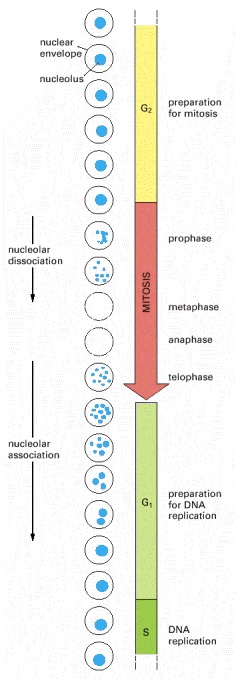
Only the cell nucleus is represented in this diagram. In most eucaryotic cells the nuclear membrane breaks down during mitosis, as indicated by the dashed circles.
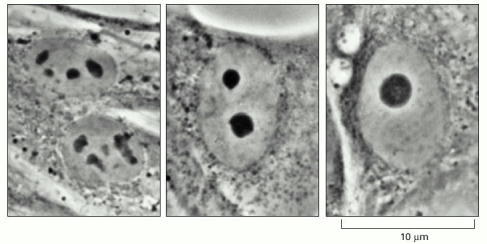
These light micrographs of human fibroblasts grown in culture show various stages of nucleolar fusion. After mitosis, each of the ten human chromosomes that carry a cluster of rRNA genes begins to form a tiny nucleolus, but these rapidly coalesce as they grow to form the single large nucleolus typical of many interphase cells. (Courtesy of E.G. Jordan and J. McGovern.)
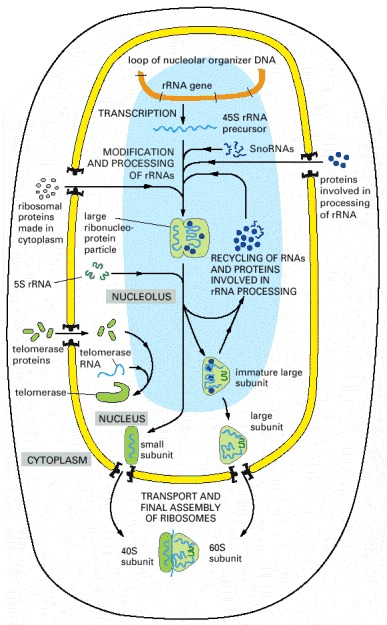
The 45S precursor rRNA is packaged in a large ribonucleoprotein particle containing many ribosomal proteins imported from the cytoplasm. While this particle remains in the nucleolus, selected pieces are added and others discarded as it is processed into immature large and small ribosomal subunits. The two ribosomal subunits are thought to attain their final functional form only as each is individually transported through the nuclear pores into the cytoplasm. Other ribonucleoprotein complexes, including telomerase shown here, are also assembled in the nucleolus.
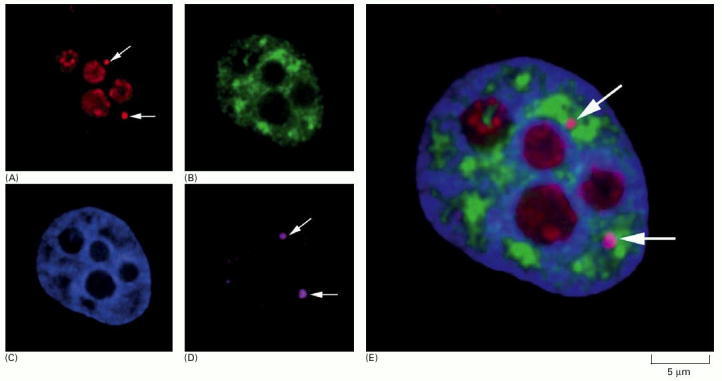
(A)-(D) show micrographs of the same human cell nucleus, each processed differently to show a particular set of nuclear structures. (E) shows an enlarged superposition of all four individual images. (A) shows the location of the protein fibrillarin (a component of several snoRNPs), which is present at both nucleoli and Cajal bodies, the latter indicated by arrows. (B) shows interchromatin granule clusters or “speckles” detected by using antibodies against a protein involved in pre-mRNA splicing. (C) is stained to show bulk chromatin. (D) shows the location of the protein coilin, which is present at Cajal bodies (indicated by arrows). (From J.R. Swedlow and A.I. Lamond, Gen. Biol. 2:1–7, 2001; micrographs courtesy of Judith Sleeman.)
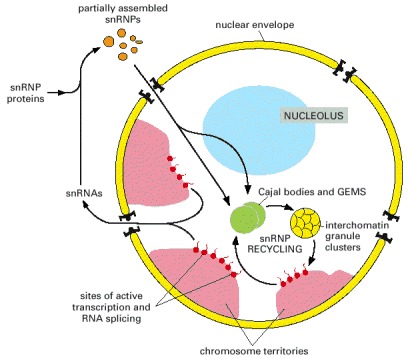
A typical vertebrate nucleus has several Cajal bodies, which are proposed to be the sites where snRNPs and snoRNPs undergo their final modifications. Interchromatin granule clusters are proposed to be storage sites for fully mature snRNPs. A typical vertebrate nucleus has 20–50 interchromatin granule clusters.
After their initial synthesis, snRNAs are exported from the nucleus, after which they undergo 5′ and 3′ end-processing and assemble with the seven common snRNP proteins (called Sm proteins). These complexes are reimported into the nucleus and the snRNPs undergo their final modification in Cajal bodies. In addition, the U6 snRNP requires chemical modification by snoRNAs in the nucleolus. The sites of active transcription and splicing (approximately 2000–3000 sites per vertebrate nucleus) correspond to the “perichromatin fibers” seen under the electron microscope. (Adapted from J.D. Lewis and D. Tollervey, Science288:1385–1389, 2000.)
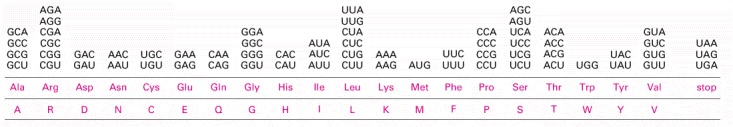
The standard one-letter abbreviation for each amino acid is presented below its three-letter abbreviation (see Panel 3-1, pp. 132–133, for the full name of each amino acid and its structure). By convention, codons are always written with the 5′-terminal nucleotide to the left. Note that most amino acids are represented by more than one codon, and that there are some regularities in the set of codons that specifies each amino acid. Codons for the same amino acid tend to contain the same nucleotides at the first and second positions, and vary at the third position. Three codons do not specify any amino acid but act as termination sites (stop codons), signaling the end of the protein-coding sequence. One codon—AUG—acts both as an initiation codon, signaling the start of a protein-coding message, and also as the codon that specifies methionine.
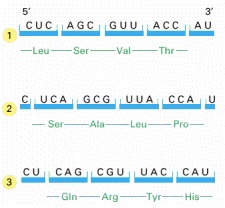
In the process of translating a nucleotide sequence (blue) into an amino acid sequence (green), the sequence of nucleotides in an mRNA molecule is read from the 5′ to the 3′ end in sequential sets of three nucleotides. In principle, therefore, the same RNA sequence can specify three completely different amino acid sequences, depending on the reading frame. In reality, however, only one of these reading frames contains the actual message.
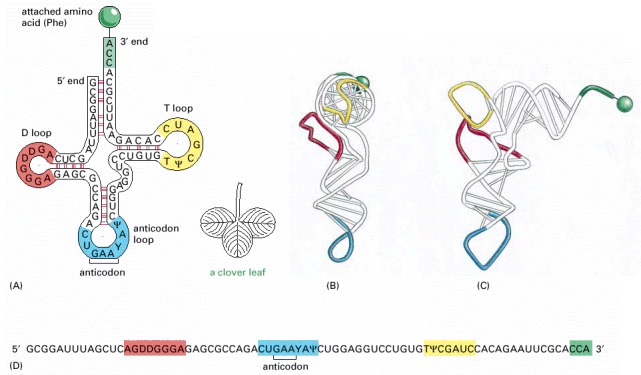
In this series of diagrams, the same tRNA molecule—in this case a tRNA specific for the amino acid phenylalanine (Phe)—is depicted in various ways. (A) The cloverleaf structure, a convention used to show the complementary base-pairing (red lines) that creates the double-helical regions of the molecule. The anticodon is the sequence of three nucleotides that base-pairs with a codon in mRNA. The amino acid matching the codon/anticodon pair is attached at the 3′ end of the tRNA. tRNAs contain some unusual bases, which are produced by chemical modification after the tRNA has been synthesized. For example, the bases denoted Ψ (for pseudouridine—see Figure 6-43) and D (for dihydrouridine—see Figure 6-55) are derived from uracil. (B and C) Views of the actual L-shaped molecule, based on x-ray diffraction analysis. Although a particular tRNA, that for the amino acid phenylalanine, is depicted, all other tRNAs have very similar structures. (D) The linear nucleotide sequence of the molecule, color-coded to match A, B, and C.
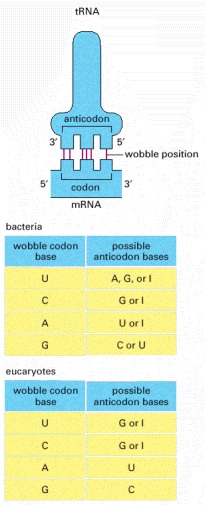
If the nucleotide listed in the first column is present at the third, or wobble, position of the codon, it can base-pair with any of the nucleotides listed in the second column. Thus, for example, when inosine (I) is present in the wobble position of the tRNA anticodon, the tRNA can recognize any one of three different codons in bacteria and either of two codons in eucaryotes. The inosine in tRNAs is formed from the deamination of guanine (see Figure 6-55), a chemical modification which takes place after the tRNA has been synthesized. The nonstandard base pairs, including those made with inosine, are generally weaker than conventional base pairs. Note that codon-anticodon base pairing is more stringent at positions 1 and 2 of the codon: here only conventional base pairs are permitted. The differences in wobble base-pairing interactions between bacteria and eucaryotes presumably result from subtle structural differences between bacterial and eucaryotic ribosomes, the molecular machines that perform protein synthesis. (Adapted from C. Guthrie and J. Abelson, in The Molecular Biology of the Yeast Saccharomyces: Metabolism and Gene Expression, pp. 487–528. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, 1982.)
The endonuclease (a four-subunit enzyme) removes the tRNA intron (blue). A second enzyme, a multifunctional tRNA ligase (not shown), then joins the two tRNA halves together. (Courtesy of Hong Li, Christopher Trotta, and John Abelson.)
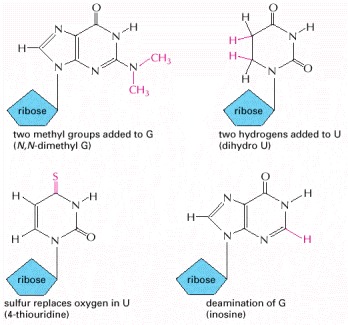
These nucleotides are produced by covalent modification of a normal nucleotide after it has been incorporated into an RNA chain. In most tRNA molecules about 10% of the nucleotides are modified (see Figure 6-52).
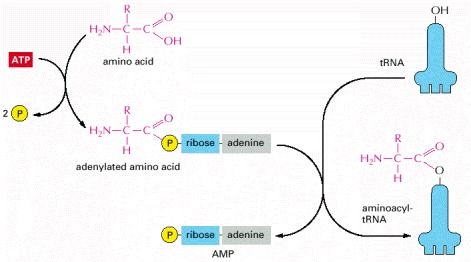
The two-step process in which an amino acid (with its side chain denoted by R) is activated for protein synthesis by an aminoacyl-tRNA synthetase enzyme is shown. As indicated, the energy of ATP hydrolysis is used to attach each amino acid to its tRNA molecule in a high-energy linkage. The amino acid is first activated through the linkage of its carboxyl group directly to an AMP moiety, forming an adenylated amino acid; the linkage of the AMP, normally an unfavorable reaction, is driven by the hydrolysis of the ATP molecule that donates the AMP. Without leaving the synthetase enzyme, the AMP-linked carboxyl group on the amino acid is then transferred to a hydroxyl group on the sugar at the 3′ end of the tRNA molecule. This transfer joins the amino acid by an activated ester linkage to the tRNA and forms the final aminoacyl-tRNA molecule. The synthetase enzyme is not shown in this diagram.
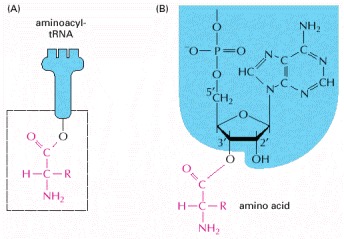
The carboxyl end of the amino acid forms an ester bond to ribose. Because the hydrolysis of this ester bond is associated with a large favorable change in free energy, an amino acid held in this way is said to be activated. (A) Schematic drawing of the structure. The amino acid is linked to the nucleotide at the 3′ end of the tRNA (see Figure 6-52). (B) Actual structure corresponding to boxed region in (A). These are two major classes of synthetase enzymes: one links the amino acid directly to the 3′-OH group of the ribose, and the other links it initially to the 2′-OH group. In the latter case, a subsequent transesterification reaction shifts the amino acid to the 3′ position. As in Figure 6-56, the “R-group” indicates the side chain of the amino acid.
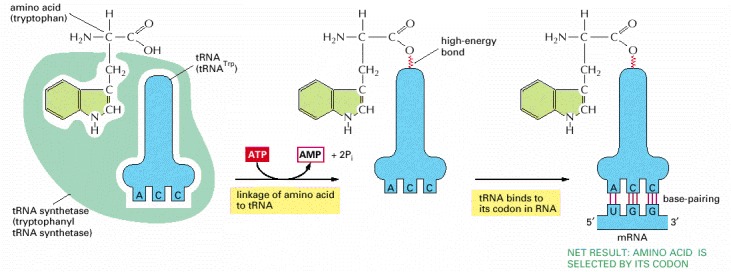
The first adaptor is the aminoacyl-tRNA synthetase, which couples a particular amino acid to its corresponding tRNA; the second adaptor is the tRNA molecule itself, whose anticodon forms base pairs with the appropriate codon on the mRNA. An error in either step would cause the wrong amino acid to be incorporated into a protein chain. In the sequence of events shown, the amino acid tryptophan (Trp) is selected by the codon UGG on the mRNA.
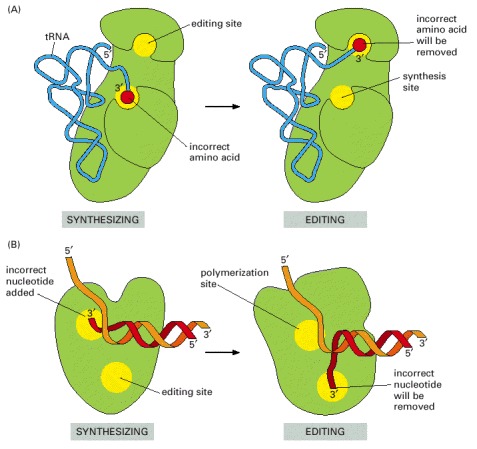
(A) tRNA synthetases remove their own coupling errors through hydrolytic editing of incorrectly attached amino acids. As described in the text, the correct amino acid is rejected by the editing site. (B) The error-correction process performed by DNA polymerase shows some similarities; however, it differs so far as the removal process depends strongly on a mispairing with the template (see Figure 5-9).
For this tRNA (tRNAGln), specific nucleotides in both the anticodon (bottom) and the amino acid-accepting arm allow the correct tRNA to be recognized by the synthetase enzyme (blue). (Courtesy of Tom Steitz.)
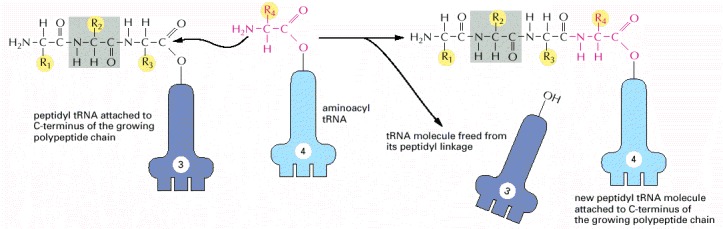
A polypeptide chain grows by the stepwise addition of amino acids to its C-terminal end. The formation of each peptide bond is energetically favorable because the growing C-terminus has been activated by the covalent attachment of a tRNA molecule. The peptidyl-tRNA linkage that activates the growing end is regenerated during each addition. The amino acid side chains have been abbreviated as R1, R2, R3, and R4; as a reference point, all of the atoms in the second amino acid in the polypeptide chain are shaded gray. The figure shows the addition of the fourth amino acid to the growing chain.
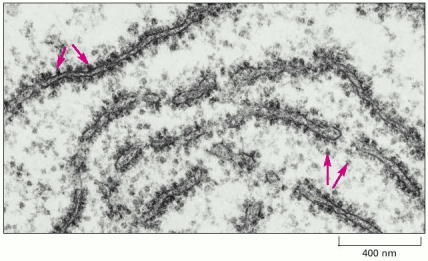
This electron micrograph shows a thin section of a small region of cytoplasm. The ribosomes appear as black dots (red arrows). Some are free in the cytosol; others are attached to membranes of the endoplasmic reticulum. (Courtesy of Daniel S. Friend.)
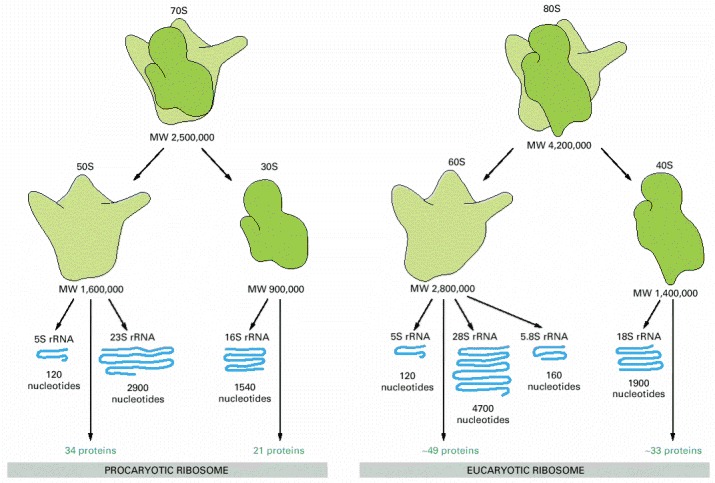
Ribosomal components are commonly designated by their “S values,” which refer to their rate of sedimentation in an ultracentrifuge. Despite the differences in the number and size of their rRNA and protein components, both procaryotic and eucaryotic ribosomes have nearly the same structure and they function similarly. Although the 18S and 28S rRNAs of the eucaryotic ribosome contain many extra nucleotides not present in their bacterial counterparts, these nucleotides are present as multiple insertions that form extra domains and leave the basic structure of each rRNA largely unchanged.
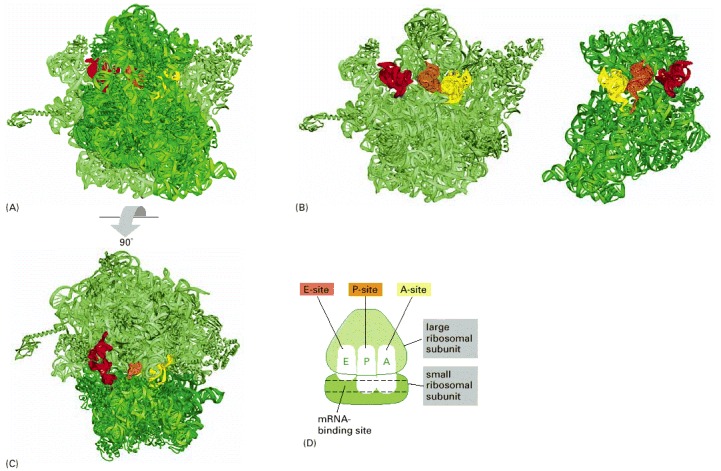
Each ribosome has three binding sites for tRNA: the A-, P-, and E-sites (short for aminoacyl-tRNA, peptidyl-tRNA, and exit, respectively) and one binding site for mRNA. (A) Structure of a bacterial ribosome with the small subunit in the front (dark green) and the large subunit in the back (light green). Both the rRNAs and the ribosomal proteins are shown. tRNAs are shown bound in the E-site (red), the P-site (orange) and the A-site (yellow). Although all three tRNA sites are shown occupied here, during the process of protein synthesis not more than two of these sites are thought to contain tRNA molecules at any one time (see Figure 6-65). (B) Structure of the large (left) and small (right) ribosomal subunits arranged as though the ribosome in (A) were opened like a book. (C) Structure of the ribosome in (A) viewed from the top. (D) Highly schematic representation of a ribosome (in the same orientation as C), which will be used in subsequent figures. (A, B, and C, adapted from M.M. Yusupov et al., Science 292:883–896, 2001, courtesy of Albion Bausom and Harry Noller.)
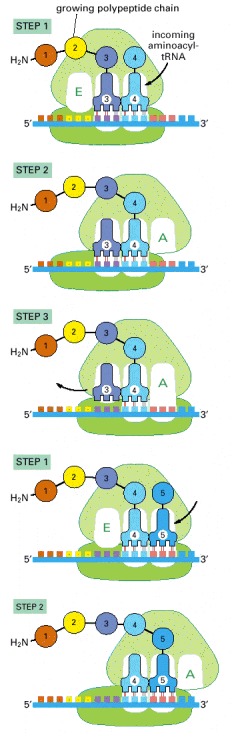
Each amino acid added to the growing end of a polypeptide chain is selected by complementary base-pairing between the anticodon on its attached tRNA molecule and the next codon on the mRNA chain. Because only one of the many types of tRNA molecules in a cell can base-pair with each codon, the codon determines the specific amino acid to be added to the growing polypeptide chain. The three-step cycle shown is repeated over and over during the synthesis of a protein. An aminoacyl-tRNA molecule binds to a vacant A-site on the ribosome in step 1, a new peptide bond is formed in step 2, and the mRNA moves a distance of three nucleotides through the small-subunit chain in step 3, ejecting the spent tRNA molecule and “resetting” the ribosome so that the next aminoacyl-tRNA molecule can bind. Although the figure shows a large movement of the small ribosome subunit relative to the large subunit, the conformational changes that actually take place in the ribosome during translation are more subtle. It is likely that they involve a series of small rearrangements within each subunit as well as several small shifts between the two subunits. As indicated, the mRNA is translated in the 5′-to-3′ direction, and the N-terminal end of a protein is made first, with each cycle adding one amino acid to the C-terminus of the polypeptide chain. The position at which the growing peptide chain is attached to a tRNA does not change during the elongation cycle: it is always linked to the tRNA present in the P site of the large subunit.
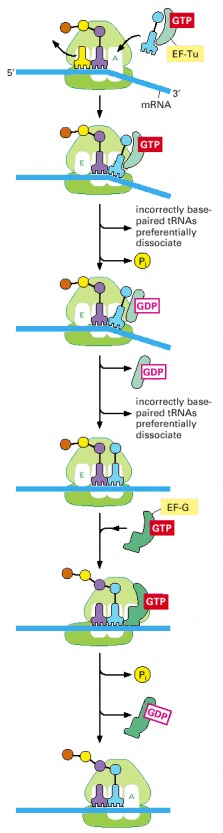
The outline of translation presented in Figure 6-65 has been supplemented with additional features, including the participation of elongation factors and a mechanism by which translational accuracy is improved. In the initial binding event (top panel) an aminoacyl-tRNA molecule that is tightly bound to EF-Tu pairs transiently with the codon at the A-site in the small subunit. During this step (second panel), the tRNA occupies a hybrid-binding site on the ribosome. The codon-anticodon pairing triggers GTP hydrolysis by EF-Tu causing it to dissociate from the aminoacyl-tRNA, which now enters the A-site (fourth panel) and can participate in chain elongation. A delay between aminoacyl-tRNA binding and its availability for protein synthesis is thereby inserted into the protein synthesis mechanism. As described in the text, this delay increases the accuracy of translation. In subsequent steps, elongation factor EF-G in the GTP-bound form enters the ribosome and binds in or near the A-site on the large ribosomal subunit, accelerating the movement of the two bound tRNAs into the A/P and P/E hybrid states. Contact with the ribosome stimulates the GTPase activity of EF-G, causing a dramatic conformational change in EF-G as it switches from the GTP to the GDP-bound form. This change moves the tRNA bound to the A/P hybrid state to the P-site and advances the cycle of translation forward by one codon.
During each cycle of translation elongation, the tRNAs molecules move through the ribosome in an elaborate series of gyrations during which they transiently occupy several “hybrid” binding states. In one, the tRNA is simultaneously bound to the A site of the small subunit and the P site of the large subunit; in another, the tRNA is bound to the P site of the small subunit and the E site of the large subunit. In a single cycle, a tRNA molecule is considered to occupy six different sites, the initial binding site (called the A/T hybrid state), the A/A site, the A/P hybrid state, the P/P site, the P/E hybrid state, and the E-site. Each tRNA is thought to ratchet through these positions, undergoing rotations along its long axis at each change in location.
EF-Tu and EF-G are the designations used for the bacterial elongation factors; in eucaryotes, they are called EF-1 and EF-2, respectively. The dramatic change in the three-dimensional structure of EF-Tu that is caused by GTP hydrolysis was illustrated in Figure 3-74. For each peptide bond formed, a molecule of EF-Tu and EF-G are each released in their inactive, GDP-bound forms. To be used again, these proteins must have their GDP exchanged for GTP. In the case of EF-Tu, this exchange is performed by a specific member of a large class of proteins known as GTP exchange factors.
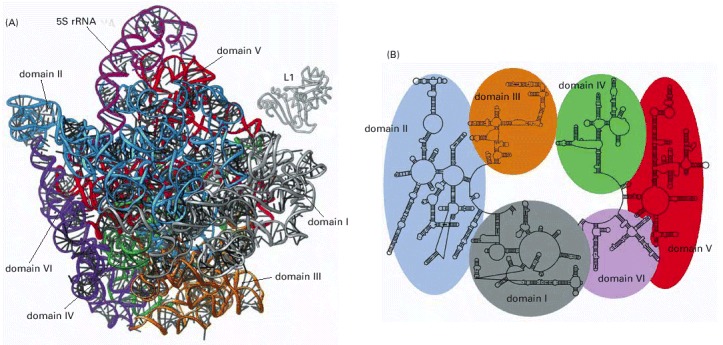
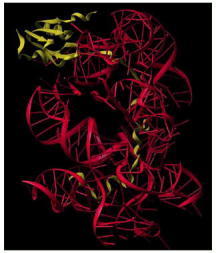
(A) Three-dimensional structures of the large-subunit rRNAs (5S and 23S) as they appear in the ribosome. One of the protein subunits of the ribosome (L1) is also shown as a reference point, since it forms a characteristic protrusion on the ribosome. (B) Schematic diagram of the secondary structure of the 23S rRNA showing the extensive network of base-pairing. The structure has been divided into six structural ‘domains’ whose colors correspond to those of the three-dimensional structure in (A). The secondary-structure diagram is highly schematized to represent as much of the structure as possible in two dimensions. To do this, several discontinuities in the RNA chain have been introduced, although in reality the 23S RNA is a single RNA molecule. For example, the base of Domain III is continuous with the base of Domain IV even though a gap appears in the diagram. (Adapted from N. Ban et al., Science 289:905–920, 2000.)
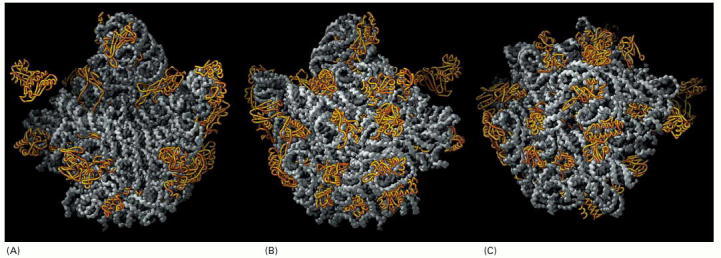
The rRNAs (5S and 23S) are depicted in gray and the large-subunit proteins (27 of the 31 total) in gold. For convenience, the protein structures depict only the polypeptide backbones. (A) View of the interface with the small subunit, the same view shown in Figure 6-64B. (B) View of the back of the large subunit, obtained by rotating (A) by 180° around a vertical axis. (C) View of the bottom of the large subunit showing the peptide exit channel in the center of the structure. (From N. Ban et al., Science 289:905–920, 2000. © AAAS.)
The globular domain of the protein lies on the surface of the ribosome and an extended region penetrates deeply into the RNA core of the ribosome. The L15 protein is shown in yellow and a portion of the ribosomal RNA core is shown in red. (Courtesy of D. Klein, P.B. Moore and T.A. Steitz.)
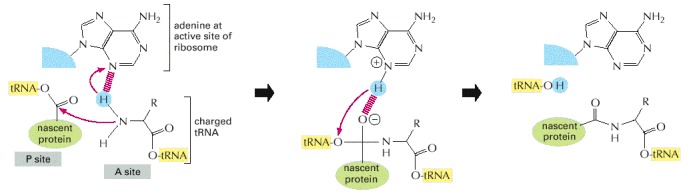
The overall reaction is catalyzed by an active site in the 23S rRNA. In the first step of the proposed mechanism, the N3 of the active-site adenine abstracts a proton from the amino acid attached to the tRNA at the ribosome's A-site, allowing its amino nitrogen to attack the carboxyl group at the end of the growing peptide chain. In the next step this protonated adenine donates its hydrogen to the oxygen linked to the peptidyl-tRNA, causing this tRNA's release from the peptide chain. This leaves a polypeptide chain that is one amino acid longer than the starting reactants. The entire reaction cycle would then repeat with the next aminoacyl tRNA that enters the A-site. (Adapted from P. Nissen et al., Science 289:920–930, 2000.)
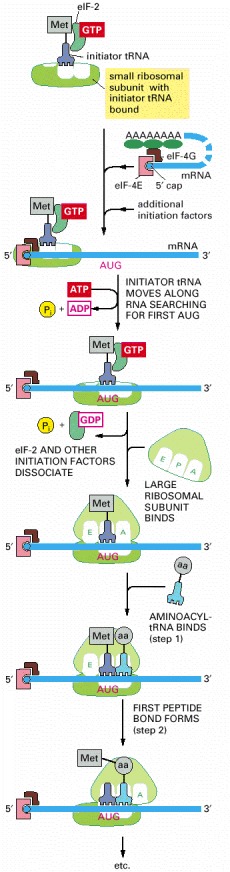
Only three of the many translation initiation factors required for this process are shown. Efficient translation initiation also requires the poly-A tail of the mRNA bound by poly-A-binding proteins which, in turn, interact with eIF4G. In this way, the translation apparatus ascertains that both ends of the mRNA are intact before initiating (see Figure 6-40). Although only one GTP hydrolysis event is shown in the figure, a second is known to occur just before the large and small ribosomal subunits join.
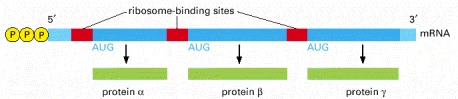
Unlike eucaryotic ribosomes, which typically require a capped 5′ end, procaryotic ribosomes initiate transcription at ribosome-binding sites (Shine-Dalgarno sequences), which can be located anywhere along an mRNA molecule. This property of ribosomes permits bacteria to synthesize more than one type of protein from a single mRNA molecule.
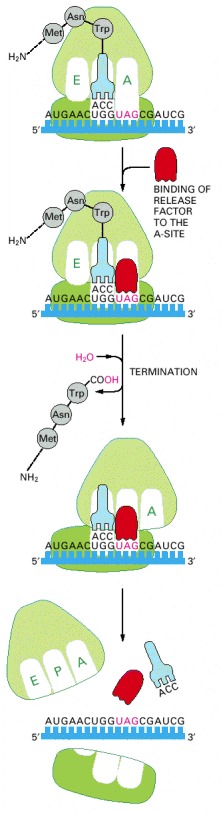
The binding of a release factor to an A-site bearing a stop codon terminates translation. The completed polypeptide is released and, after the action of a ribosome recycling factor (not shown), the ribosome dissociates into its two separate subunits.
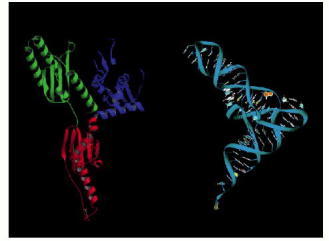
The protein is on the left and the tRNA on the right. (From H. Song et al., Cell 100:311–321, 2000. © Elsevier.)
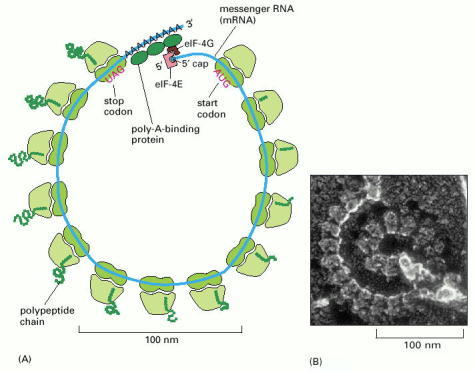
(A) Schematic drawing showing how a series of ribosomes can simultaneously translate the same eucaryotic mRNA molecule. (B) Electron micrograph of a polyribosome from a eucaryotic cell. (B, courtesy of John Heuser.)
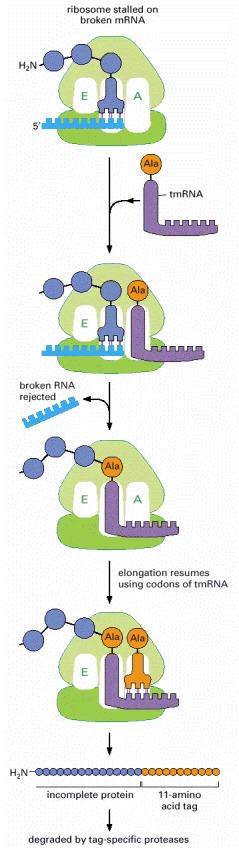
The tmRNA shown is a 363-nucleotide RNA with both tRNA and mRNA functions, hence its name. It carries an alanine and can enter the vacant A-site of a stalled ribosome to add this alanine to a polypeptide chain, mimicking a tRNA except that no codon is present to guide it. The ribosome then translates ten codons from the tmRNA, completing an 11-amino acid tag on the protein. This tag is recognized by proteases that then degrade the entire protein.
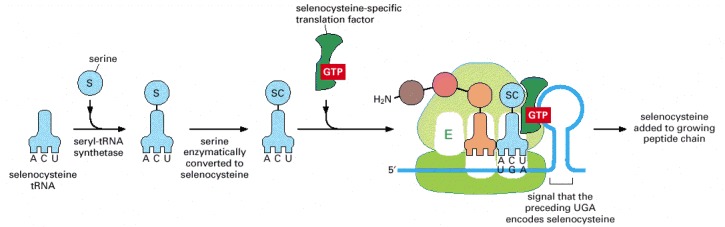
A specialized tRNA is charged with serine by the normal seryl-tRNA synthetase, and the serine is subsequently converted enzymatically to selenocysteine. A specific RNA structure in the mRNA (a stem and loop structure with a particular nucleotide sequence) signals that selenocysteine is to be inserted at the neighboring UGA codon. As indicated, this event requires the participation of a selenocysteine-specific translation factor.
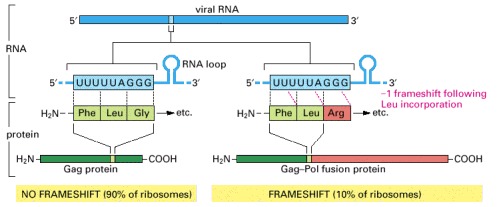
The viral reverse transcriptase and integrase are produced by proteolytic processing of a large protein (the Gag-Pol fusion protein) consisting of both the Gag and Pol amino acid sequences. The viral capsid proteins are produced by proteolytic processing of the more abundant Gag protein. Both the Gag and the Gag-Pol fusion proteins start identically, but the Gag protein terminates at an in-frame stop codon (not shown); the indicated frameshift bypasses this stop codon, allowing the synthesis of the longer Gag-Pol fusion protein. The frameshift occurs because features in the local RNA structure (including the RNA loop shown) cause the tRNALeu attached to the C-terminus of the growing polypeptide chain occasionally to slip backward by one nucleotide on the ribosome, so that it pairs with a UUU codon instead of the UUA codon that had initially specified its incorporation; the next codon (AGG) in the new reading frame specifies an arginine rather than a glycine. This controlled slippage is due in part to a stem and loop structure that forms in the viral mRNA, as indicated in the figure. The sequence shown is from the human AIDS virus, HIV. (Adapted from T. Jacks et al., Nature 331:280–283, 1988.)
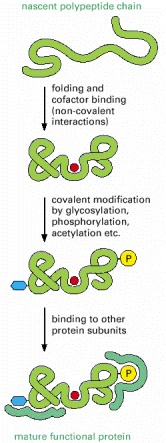
As indicated, translation of an mRNA sequence into an amino acid sequence on the ribosome is not the end of the process of forming a protein. To be useful to the cell, the completed polypeptide chain must fold correctly into its three-dimensional conformation, bind any cofactors required, and assemble with its partner protein chains (if any). These changes are driven by noncovalent bond formation. As indicated, many proteins also have covalent modifications made to selected amino acids. Although the most frequent of these are protein glycosylation and protein phosphorylation, more than 100 different types of covalent modifications are known (see, for example, Figure 4-35).
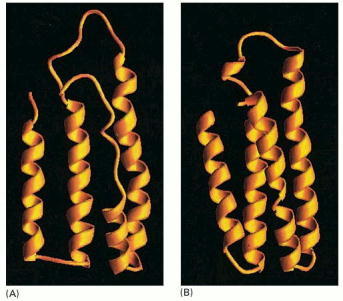
(A) A molten globule form of cytochrome b562 is more open and less highly ordered than the final folded form of the protein, shown in (B). Note that the molten globule contains most of the secondary structure of the final form, although the ends of the α helices are frayed and one of the helices is only partly formed. (Courtesy of Joshua Wand, from Y. Feng et al., Nat. Struct. Biol. 1:30–35, 1994.)
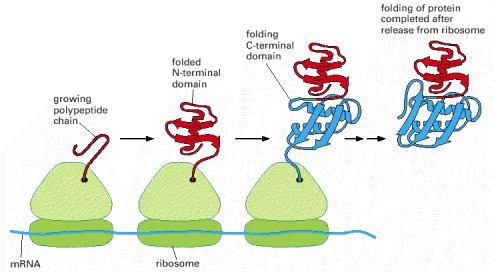
A growing polypeptide chain is shown acquiring its secondary and tertiary structure as it emerges from a ribosome. The N-terminal domain folds first, while the C-terminal domain is still being synthesized. In this case, the protein has not yet achieved its final conformation by the time it is released from the ribosome. (Modified from A.N. Federov and T.O. Baldwin, J. Biol. Chem. 272:32715–32718, 1997.)
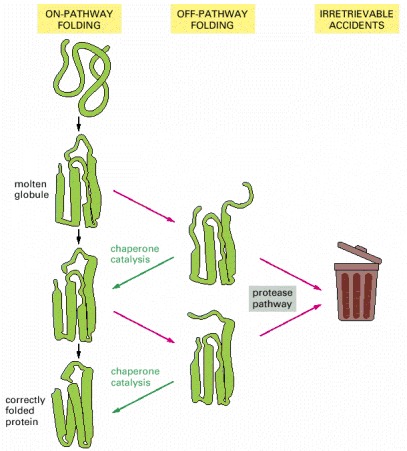
Each domain of a newly synthesized protein rapidly attains a “molten globule” state. Subsequent folding occurs more slowly and by multiple pathways, often involving the help of a molecular chaperone. Some molecules may still fail to fold correctly; as explained shortly, these are recognized and degraded by specific proteases.
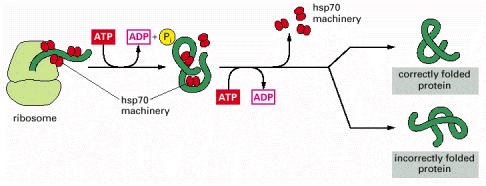
These proteins act early, recognizing a small stretch of hydrophobic amino acids on a protein's surface. Aided by a set of smaller hsp40 proteins, an hsp70 monomer binds to its target protein and then hydrolyzes a molecule of ATP to ADP, undergoing a conformational change that causes the hsp70 to clamp down very tightly on the target. After the hsp40 dissociates, the dissociation of the hsp70 protein is induced by the rapid re-binding of ATP after ADP release. Repeated cycles of hsp protein binding and release help the target protein to refold, as schematically illustrated in Figure 6-82.
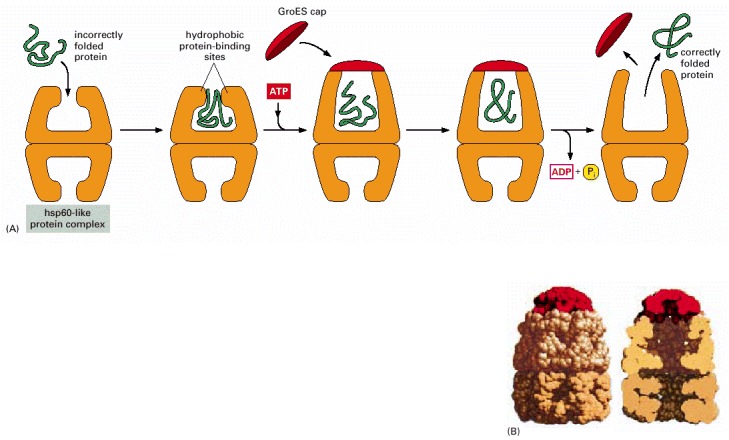
(A) The catalysis of protein refolding. As indicated, a misfolded protein is initially captured by hydrophobic interactions along one rim of the barrel. The subsequent binding of ATP plus a protein cap increases the diameter of the barrel rim, which may transiently stretch (partly unfold) the client protein. This also confines the protein in an enclosed space, where it has a new opportunity to fold. After about 15 seconds, ATP hydrolysis ejects the protein, whether folded or not, and the cycle repeats. This type of molecular chaperone is also known as a chaperonin; it is designated as hsp60 in mitochondria, TCP-1 in the cytosol of vertebrate cells, and GroEL in bacteria. As indicated, only half of the symmetrical barrel operates on a client protein at any one time. (B) The structure of GroEL bound to its GroES cap, as determined by x-ray crystallography. On the left is shown the outside of the barrel-like structure and on the right a cross section through its center. (B, adapted from B. Bukace and A.L. Horwich, Cell 92:351–366, 1998.)
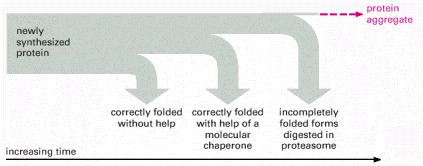
As indicated, a newly synthesized protein sometimes folds correctly and assembles with its partners on its own, in which case it is left alone. Incompletely folded proteins are helped to refold by molecular chaparones: first by a family of hsp70 proteins, and if this fails, then by hsp60-like proteins. In both cases the client proteins are recognized by an abnormally exposed patch of hydrophobic amino acids on their surface. These processes compete with a different system that recognizes an abnormally exposed patch and transfers the protein that contains it to a proteasome for complete destruction. The combination of all of these processes is needed to prevent massive protein aggregation in a cell, which can occur when many hydrophobic regions on proteins clump together and precipitate the entire mass out of solution.
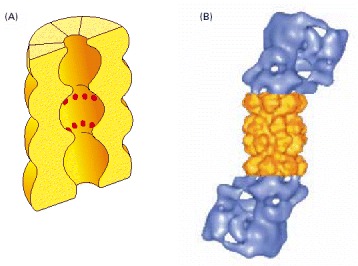
(A) A cut-away view of the structure of the central 20S cylinder, as determined by x-ray crystallography, with the active sites of the proteases indicated by red dots. (B) The structure of the entire proteasome, in which the central cylinder (yellow) is supplemented by a 19S cap (blue) at each end, whose structure has been determined by computer processing of electron microscope images. The complex cap structure selectively binds those proteins that have been marked for destruction; it then uses ATP hydrolysis to unfold their polypeptide chains and feed them into the inner chamber of the 20S cylinder for digestion to short peptides. (B, from W. Baumeister et al., Cell 92:367–380, 1998. © Elsevier.)
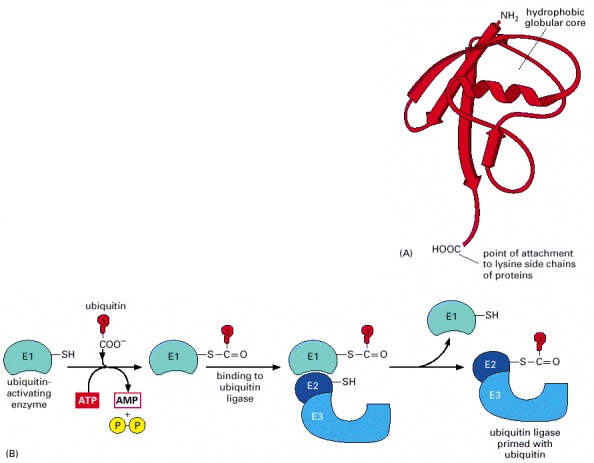
(A) The three-dimensional structure of ubiquitin; this relatively small protein contains 76 amino acids. (B) The C-terminus of ubiquitin is initially activated through its high-energy thioester linkage to a cysteine side chain on the E1 protein. This reaction requires ATP, and it proceeds via a covalent AMP-ubiquitin intermediate. The activated ubiquitin on E1, also known as the ubiquitin-activating enzyme, is then transferred to the cysteines on a set of E2 molecules. These E2s exist as complexes with an even larger family of E3 molecules. (C) The addition of a multiubiquitin chain to a target protein. In a mammalian cell there are roughly 300 distinct E2-E3 complexes, each of which recognizes a different degradation signal on a target protein by means of its E3 component. The E2s are called ubiquitin-conjugating enzymes. The E3s have been referred to traditionally as ubiquitin ligases, but it is more accurate to reserve this name for the functional E2-E3 complex.
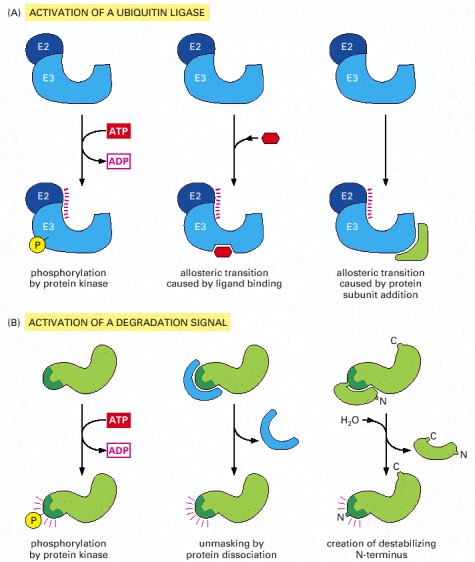
(A) Activation of a specific E3 molecule creates a new ubiquitin ligase. (B) Creation of an exposed degradation signal in the protein to be degraded. This signal binds a ubiquitin ligase, causing the addition of a multiubiquitin chain to a nearby lysine on the target protein. All six pathways shown are known to be used by cells to induce the movement of selected proteins into the proteasome.
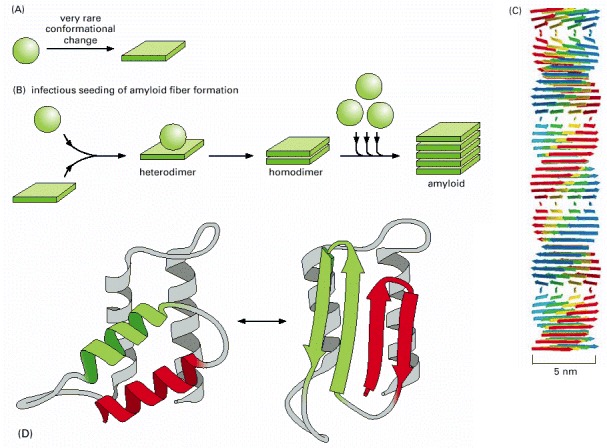
(A) Schematic illustration of the type of conformational change in a protein that produces material for a cross-beta filament. (B) Diagram illustrating the self-infectious nature of the protein aggregation that is central to prion diseases. PrP is highly unusual because the misfolded version of the protein, called PrP*, induces the normal PrP protein it contacts to change its conformation, as shown. Most of the human diseases caused by protein aggregation are caused by the overproduction of a variant protein that is especially prone to aggregation, but because this structure is not infectious in this way, it cannot spread from one animal to another. (C) Drawing of a cross-beta filament, a common type of protease-resistant protein aggregate found in a variety of human neurological diseases. Because the hydrogen-bond interactions in a β sheet form between polypeptide backbone atoms (see Figure 3-9), a number of different abnormally folded proteins can produce this structure. (D) One of several possible models for the conversion of PrP to PrP*, showing the likely change of two α-helices into four β-strands. Although the structure of the normal protein has been determined accurately, the structure of the infectious form is not yet known with certainty because the aggregation has prevented the use of standard structural techniques. (C, courtesy of Louise Serpell, adapted from M. Sunde et al., J. Mol. Biol. 273:729–739, 1997; D, adapted from S.B. Prusiner, Trends Biochem. Sci. 21:482–487, 1996.)
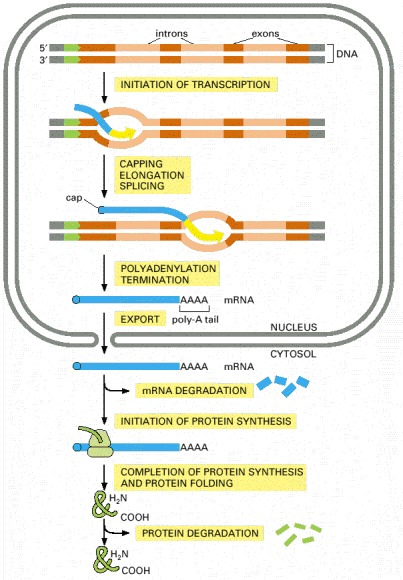
The final level of each protein in a eucaryotic cell depends upon the efficiency of each step depicted.

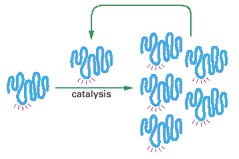
This hypothetical process would require catalysis of the production of both a second RNA strand of complementary nucleotide sequence and the use of this second RNA molecule as a template to form many molecules of RNA with the original sequence. The red rays represent the active site of this hypothetical RNA enzyme.
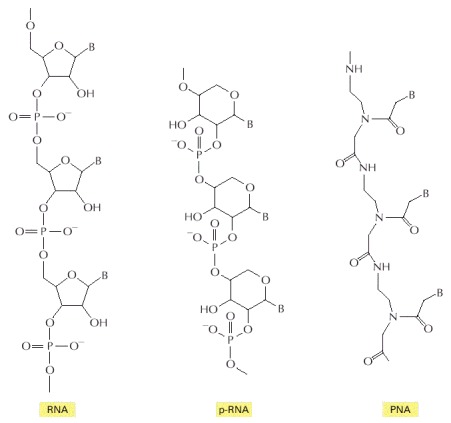
In each case, B indicates the positions of purine and pyrimidine bases. The polymer p-RNA (pyranosyl-RNA) is RNA in which the furanose (five-membered ring) form of ribose has been replaced by the pyranose (six-membered ring) form. In PNA (peptide nucleic acid), the ribose phosphate backbone of RNA has been replaced by the peptide backbone found in proteins. Like RNA, both p-RNA and PNA can form double helices through complementary base-pairing, and each could therefore in principle serve as a template for its own synthesis (see Figure 6-92).
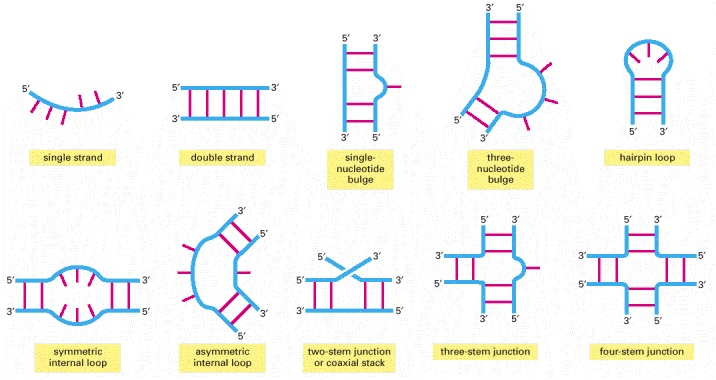
Conventional, complementary base-pairing interactions are indicated by red “rungs” in double-helical portions of the RNA.
Some of these interactions can join distant parts of the same RNA molecule or bring two separate RNA molecules together.
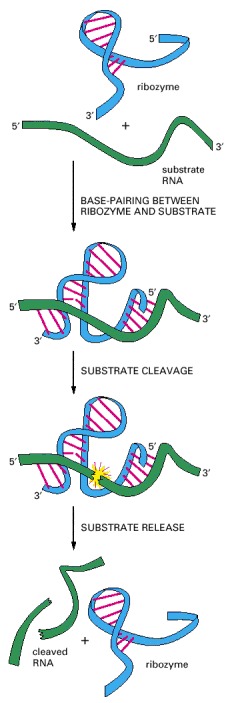
This simple RNA molecule catalyzes the cleavage of a second RNA at a specific site. This ribozyme is found embedded in larger RNA genomes—called viroids—which infect plants. The cleavage, which occurs in nature at a distant location on the same RNA molecule that contains the ribozyme, is a step in the replication of the viroid genome. Although not shown in the figure, the reaction requires a molecule of Mg positioned at the active site. (Adapted from T.R. Cech and O.C. Uhlenbeck, Nature 372:39–40, 1994.)
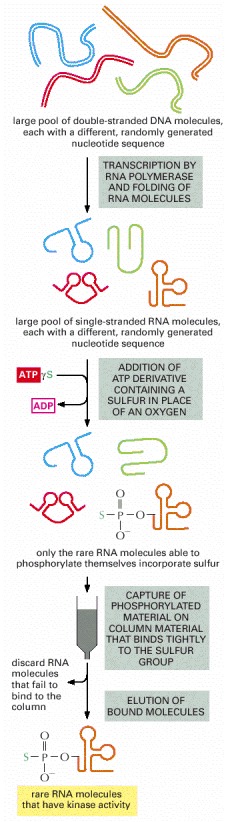
Beginning with a large pool of nucleic acid molecules synthesized in the laboratory, those rare RNA molecules that possess a specified catalytic activity can be isolated and studied. Although a specific example (that of an autophosphorylating ribozyme) is shown, variations of this procedure have been used to generate many of the ribozymes listed in Table 6-4. During the autophosphorylation step, the RNA molecules are sufficiently dilute to prevent the “cross”-phosphorylation of additional RNA molecules. In reality, several repetitions of this procedure are necessary to select the very rare RNA molecules with catalytic activity. Thus the material initially eluted from the column is converted back into DNA, amplified many fold (using reverse transcriptase and PCR as explained in Chapter 8), transcribed back into RNA, and subjected to repeated rounds of selection. (Adapted from J.R. Lorsch and J.W. Szostak, Nature 371:31–36, 1994.)
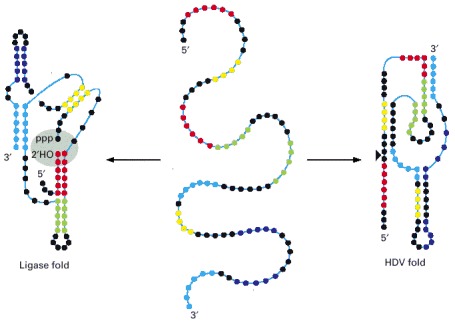
This 88-nucleotide RNA, created in the laboratory, can fold into a ribozyme that carries out a self-ligation reaction (left) or a self-cleavage reaction (right). The ligation reaction forms a 2′,5′ phosphodiester linkage with the release of pyrophosphate. This reaction seals the gap (gray shading), which was experimentally introduced into the RNA molecule. In the reaction carried out by the HDV fold, the RNA is cleaved at this same position, indicated by the arrowhead. This cleavage resembles that used in the life cycle of HDV, a hepatitis B satellite virus, hence the name of the fold. Each nucleotide is represented by a colored dot, with the colors used simply to clarify the two different folding patterns. The folded structures illustrate the secondary structures of the two ribozymes with regions of base-pairing indicated by close oppositions of the colored dots. Note that the two ribozyme folds have no secondary structure in common. (Adapted from E.A. Schultes and D.P. Bartel, Science 289:448–452, 2000.)
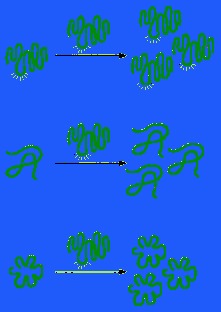
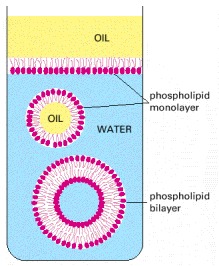
Because these molecules have hydrophilic heads and lipophilic tails, they align themselves at an oil/water interface with their heads in the water and their tails in the oil. In the water they associate to form closed bilayer vesicles in which the lipophilic tails are in contact with one another and the hydrophilic heads are exposed to the water.
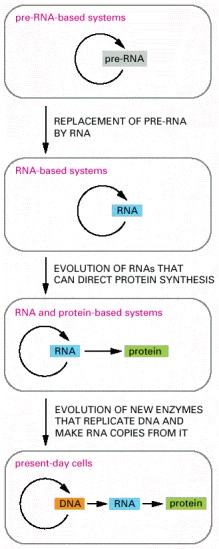
In the earliest cells, pre-RNA molecules would have had combined genetic, structural, and catalytic functions and these functions would have gradually been replaced by RNA. In present-day cells, DNA is the repository of genetic information, and proteins perform the vast majority of catalytic functions in cells. RNA primarily functions today as a go-between in protein synthesis, although it remains a catalyst for a number of crucial reactions.