Chapter 5 - Figures
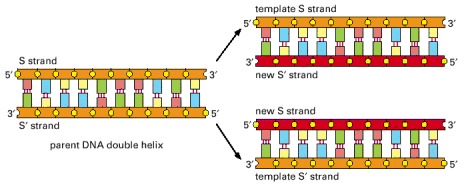
Because the nucleotide A will successfully pair only with T, and G only with C, each strand of DNA can serve as a template to specify the sequence of nucleotides in its complementary strand by DNA base-pairing. In this way, a double-helical DNA molecule can be copied precisely.
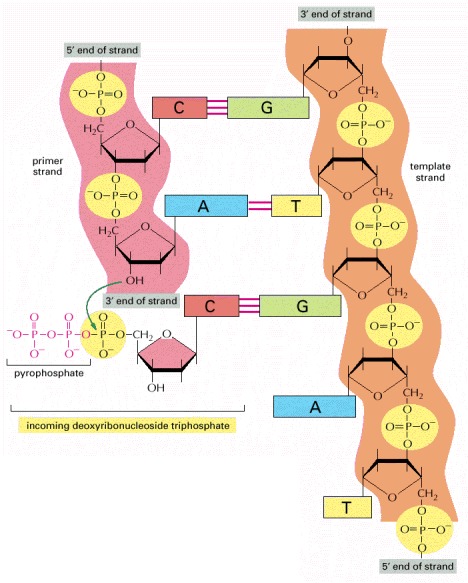
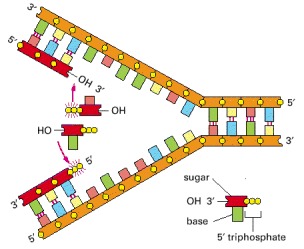
The addition of a deoxyribonucleotide to the 3′ end of a polynucleotide chain (the primer strand) is the fundamental reaction by which DNA is synthesized. As shown, base-pairing between an incoming deoxyribonucleoside triphosphate and an existing strand of DNA (the template strand) guides the formation of the new strand of DNA and causes it to have a complementary nucleotide sequence.
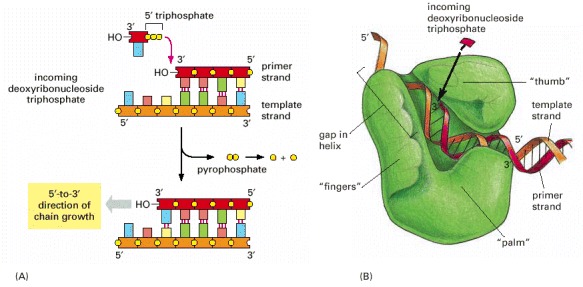
(A) As indicated, DNA polymerase catalyzes the stepwise addition of a deoxyribonucleotide to the 3′-OH end of a polynucleotide chain, the primer strand, that is paired to a second template strand. The newly synthesized DNA strand therefore polymerizes in the 5′-to-3′ direction as shown in the previous figure. Because each incoming deoxyribonucleoside triphosphate must pair with the template strand to be recognized by the DNA polymerase, this strand determines which of the four possible deoxyribonucleotides (A, C, G, or T) will be added. The reaction is driven by a large, favorable free-energy change, caused by the release of pyrophosphate and its subsequent hydrolysis to two molecules of inorganic phosphate. (B) The structure of an E. coli DNA polymerase molecule, as determined by x-ray crystallography. Roughly speaking, it resembles a right hand in which the palm, fingers, and thumb grasp the DNA. This drawing illustrates a DNA polymerase that functions during DNA repair, but the enzymes that replicate DNA have similar features. (B, adapted from L.S. Beese, V. Derbyshire, and T.A. Steitz, Science 260:352–355, 1993.)
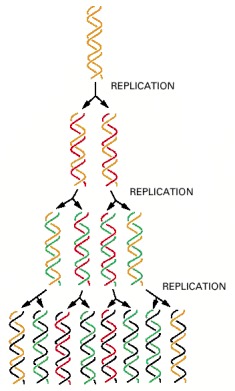
In a round of replication, each of the two strands of DNA is used as a template for the formation of a complementary DNA strand. The original strands therefore remain intact through many cell generations.
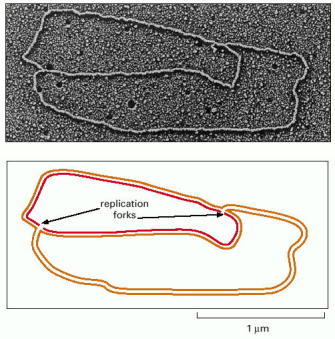
An active zone of DNA replication moves progressively along a replicating DNA molecule, creating a Y-shaped DNA structure known as a replication fork: the two arms of each Y are the two daughter DNA molecules, and the stem of the Y is the parental DNA helix. In this diagram, parental strands are orange; newly synthesized strands are red. (Micrograph courtesy of Jerome Vinograd.)
Although it might seem to be the simplest possible model for DNA replication, the mechanism illustrated here is not the one that cells use. In this scheme, both daughter DNA strands would grow continuously, using the energy of hydrolysis of the two terminal phosphates (yellow circles highlighted by red rays) to add the next nucleotide on each strand. This would require chain growth in both the 5′-to-3′ direction (top) and the 3′-to-5′ direction (bottom). No enzyme that catalyzes 3′-to-5′ nucleotide polymerization has ever been found.
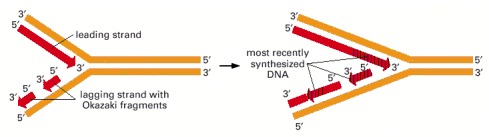
Because both daughter DNA strands are polymerized in the 5′-to-3′ direction, the DNA synthesized on the lagging strand must be made initially as a series of short DNA molecules, called Okazaki fragments.
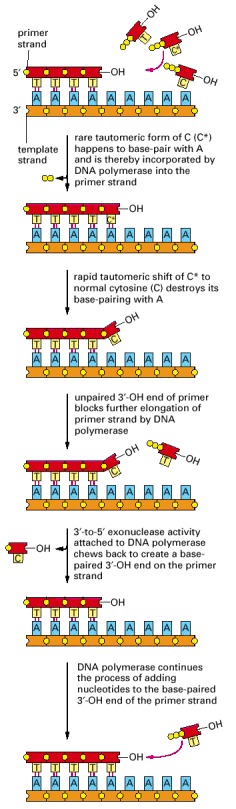
In this example, the mismatch is due to the incorporation of a rare, transient tautomeric form of C, indicated by an asterisk. But the same proofreading mechanism applies to any misincorporation at the growing 3′-OH end.
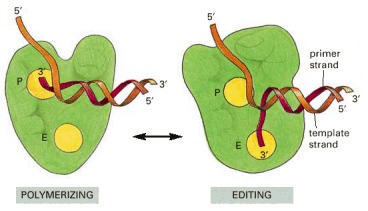
Outline of the structures of DNA polymerase complexed with the DNA template in the polymerizing mode (left) and the editing mode (right). The catalytic site for the exonucleolytic (E) and the polymerization (P) reactions are indicated. To determine these structures by x-ray crystallography, researchers “froze” the polymerases in these two states, by using either a mutant polymerase defective in the exonucleolytic domain (right), or by withholding the Mg2+ required for polymerization (left).
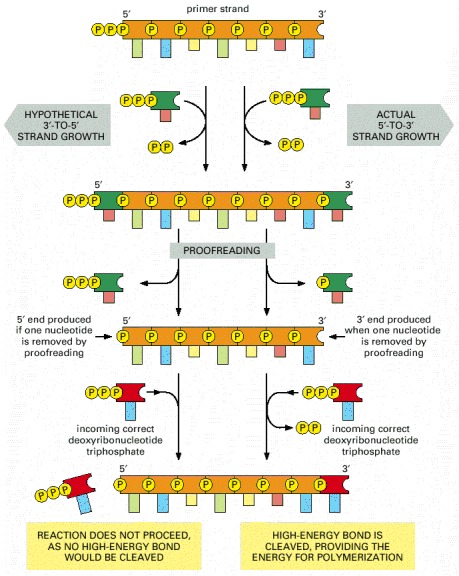
Growth in the 5′-to-3′ direction, shown on the right, allows the chain to continue to be elongated when a mistake in polymerization has been removed by exonucleolytic proofreading (see Figure 5-9). In contrast, exonucleolytic proofreading in the hypothetical 3′-to-5′ polymerization scheme, shown on the left, would block further chain elongation. For convenience, only the primer strand of the DNA double helix is shown.
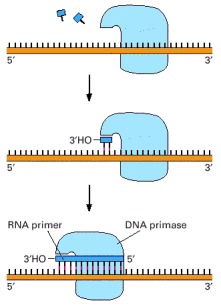
A schematic view of the reaction catalyzed by DNA primase, the enzyme that synthesizes the short RNA primers made on the lagging strand using DNA as a template. Unlike DNA polymerase, this enzyme can start a new polynucleotide chain by joining two nucleoside triphosphates together. The primase synthesizes a short polynucleotide in the 5′-to-3′ direction and then stops, making the 3′ end of this primer available for the DNA polymerase.
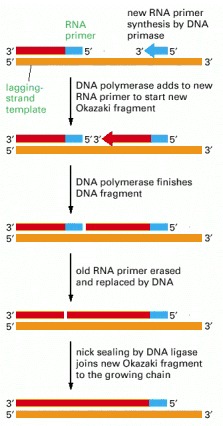
In eucaryotes, RNA primers are made at intervals spaced by about 200 nucleotides on the lagging strand, and each RNA primer is approximately 10 nucleotides long. This primer is erased by a special DNA repair enzyme (an RNAse H) that recognizes an RNA strand in an RNA/DNA helix and fragments it; this leaves gaps that are filled in by DNA polymerase and DNA ligase.
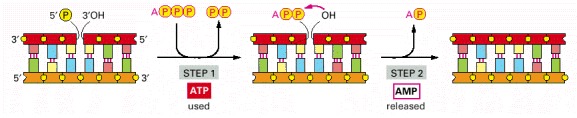
This enzyme seals a broken phosphodiester bond. As shown, DNA ligase uses a molecule of ATP to activate the 5′ end at the nick (step 1) before forming the new bond (step 2). In this way, the energetically unfavorable nick-sealing reaction is driven by being coupled to the energetically favorable process of ATP hydrolysis.
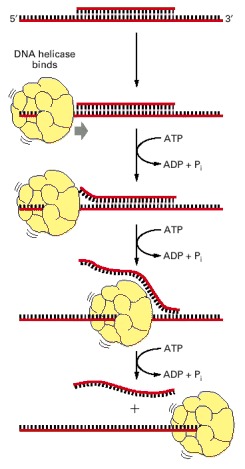
A short DNA fragment is annealed to a long DNA single strand to form a region of DNA double helix. The double helix is melted as the helicase runs along the DNA single strand, releasing the short DNA fragment in a reaction that requires the presence of both the helicase protein and ATP. The rapid step-wise movement of the helicase is powered by its ATP hydrolysis (see Figure 3-76).
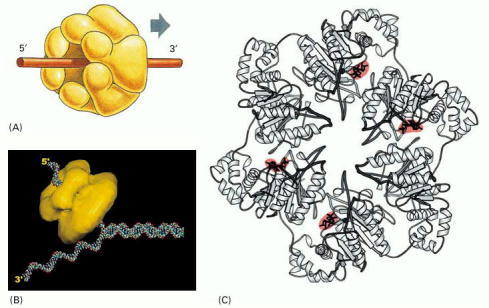
(A) A schematic diagram of the protein as a hexameric ring. (B) Schematic diagram showing a DNA replication fork and helicase to scale. (C) Detailed structure of the bacteriophage T7 replicative helicase, as determined by x-ray diffraction. Six identical subunits bind and hydrolyze ATP in an ordered fashion to propel this molecule along a DNA single strand that passes through the central hole. Red indicates bound ATP molecules in the structure. (B, courtesy of Edward H. Egelman; C, from M.R. Singleton et al., Cell 101:589–600, 2000. © Elsevier.)
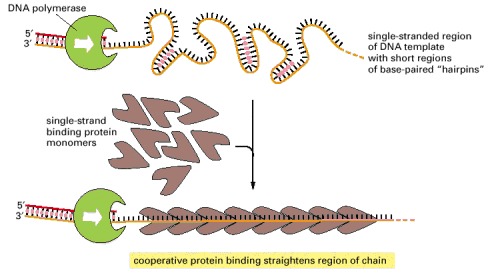
Because each protein molecule prefers to bind next to a previously bound molecule, long rows of this protein form on a DNA single strand. This cooperative binding straightens out the DNA template and facilitates the DNA polymerization process. The “hairpin helices” shown in the bare, single-stranded DNA result from a chance matching of short regions of complementary nucleotide sequence; they are similar to the short helices that typically form in RNA molecules (see Figure 1-6).
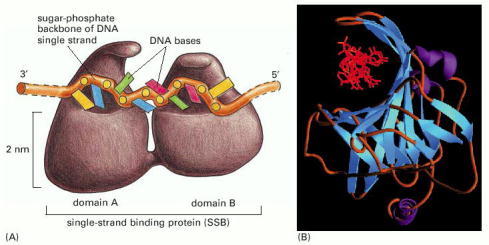
(A) A front view of the two DNA binding domains of RPA protein, which cover a total of eight nucleotides. Note that the DNA bases remain exposed in this protein–DNA complex. (B) A diagram showing the three-dimensional structure, with the DNA strand (red) viewed end-on. (B, from A. Bochkarev et al., Nature 385:176–181, 1997. © Macmillan Magazines Ltd.)
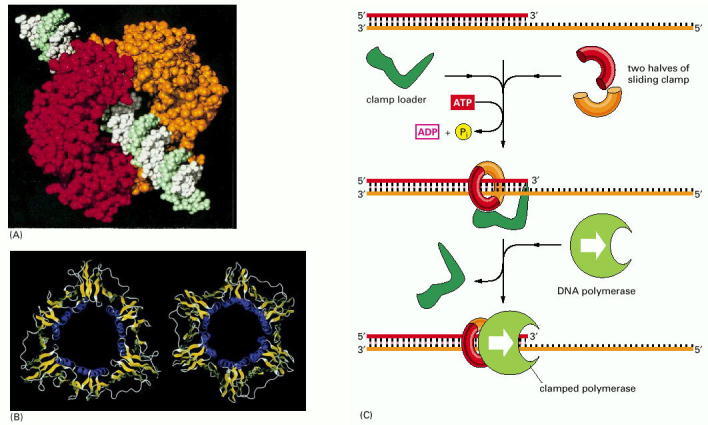
(A) The structure of the clamp protein from E. coli, as determined by x-ray crystallography, with a DNA helix added to indicate how the protein fits around DNA. (B) A similar protein is present in eucaryotes, as illustrated by this comparison of the E. coli sliding clamp (left) with the PCNA protein from humans (right). (C) Schematic illustration showing how the clamp is assembled to hold a moving DNA polymerase molecule on the DNA. In the simplified reaction shown here, the clamp loader dissociates into solution once the clamp has been assembled. At a true replication fork, the clamp loader remains close to the lagging-strand polymerase, ready to assemble a new clamp at the start of each new Okazaki fragment (see Figure 5-22). (A and B, from X.-P. Kong et al., Cell 69:425–437, 1992. © Elsevier.)
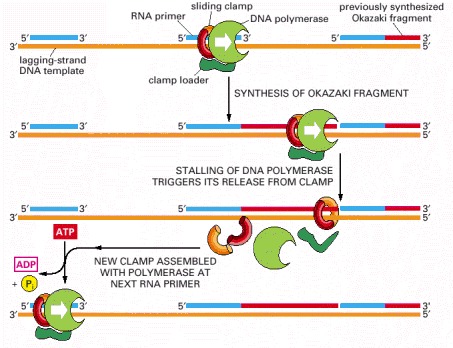
The association of the clamp loader with the lagging-strand polymerase shown here is for illustrative purposes only; in reality, the clamp loader is carried along with the replication fork in a complex that includes both the leading-strand and lagging-strand DNA polymerases (see Figure 5-22).
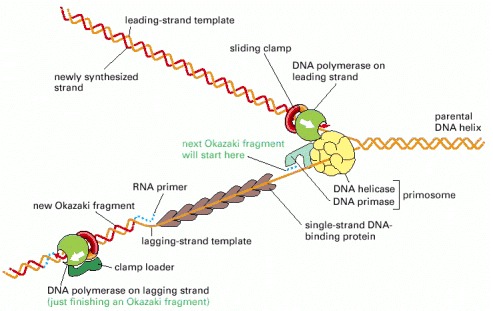
The major types of proteins that act at a DNA replication fork are illustrated, showing their approximate positions on the DNA.
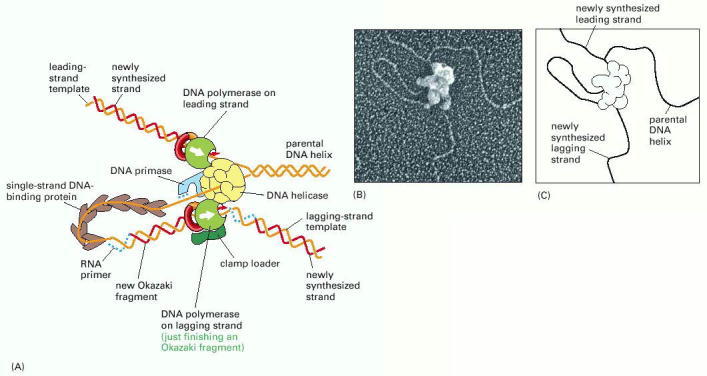
(A) This schematic diagram shows a current view of the arrangement of replication proteins at a replication fork when the fork is moving. The diagram in Figure 5-21 has been altered by folding the DNA on the lagging strand to bring the lagging-strand DNA polymerase molecule into a complex with the leading-strand DNA polymerase molecule. This folding process also brings the 3′ end of each completed Okazaki fragment close to the start site for the next Okazaki fragment (compare with Figure 5-21). Because the lagging-strand DNA polymerase molecule remains bound to the rest of the replication proteins, it can be reused to synthesize successive Okazaki fragments. In this diagram, it is about to let go of its completed DNA fragment and move to the RNA primer that will be synthesized nearby, as required to start the next DNA fragment. Note that one daughter DNA helix extends toward the bottom right and the other toward the top left in this diagram. Additional proteins help to hold the different protein components of the fork together, enabling them to function as a well-coordinated protein machine. The actual protein complex is more compact than indicated, and the clamp loader is held in place by interactions not shown here. (B) An electron micrograph showing the replication machine from the bacteriophage T4 as it moves along a template synthesizing DNA behind it. (C) An interpretation of the micrograph is given in the sketch: note especially the DNA loop on the lagging strand. Apparently, the replication proteins became partly detached from the very front of the replication fork during the preparation of this sample for electron microscopy. (B, courtesy of Jack Griffith.)
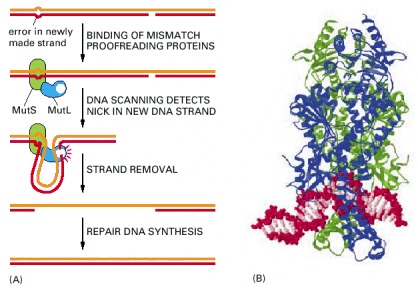
(A) The two proteins shown are present in both bacteria and eucaryotic cells: MutS binds specifically to a mismatched base pair, while MutL scans the nearby DNA for a nick. Once a nick is found, MutL triggers the degradation of the nicked strand all the way back through the mismatch. Because nicks are largely confined to newly replicated strands in eucaryotes, replication errors are selectively removed. In bacteria, the mechanism is the same, except that an additional protein in the complex (MutH) nicks unmethylated (and therefore newly replicated) GATC sequences, thereby beginning the process illustrated here. (B) The structure of the MutS protein bound to a DNA mismatch. This protein is a dimer, which grips the DNA double helix as shown, kinking the DNA at the mismatched base pair. It seems that the MutS protein scans the DNA for mismatches by testing for sites that can be readily kinked, which are those without a normal complementary base pair. (B, from G. Obmolova et al., Nature 407:703–710, 2000. © Macmillan Magazines Ltd.)
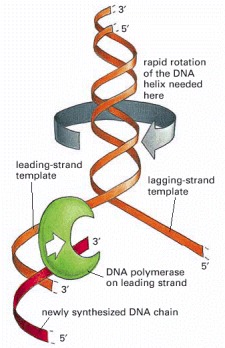
For a bacterial replication fork moving at 500 nucleotides per second, the parental DNA helix ahead of the fork must rotate at 50 revolutions per second.
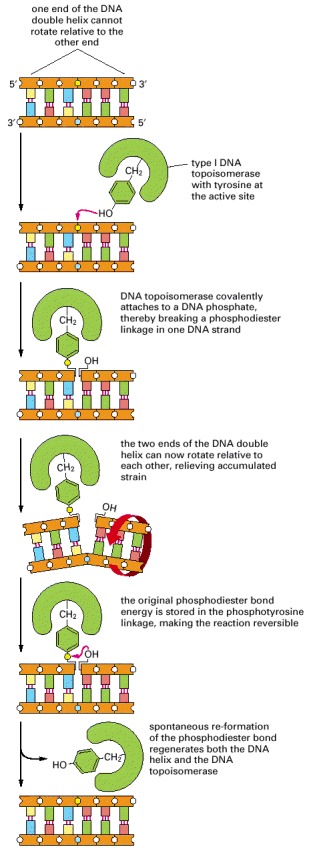
As indicated, these enzymes transiently form a single covalent bond with DNA; this allows free rotation of the DNA around the covalent backbone bonds linked to the blue phosphate.
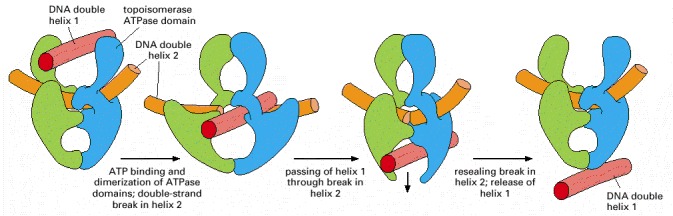
As indicated, ATP binding to the two ATPase domains causes them to dimerize and drives the reactions shown. Because a single cycle of this reaction can occur in the presence of a non-hydrolyzable ATP analog, ATP hydrolysis is thought to be needed only to reset the enzyme for each new reaction cycle. This model is based on structural and mechanistic studies of the enzyme. (Modified from J.M. Berger, Curr. Opin. Struct. Biol. 8:26–32, 1998.)
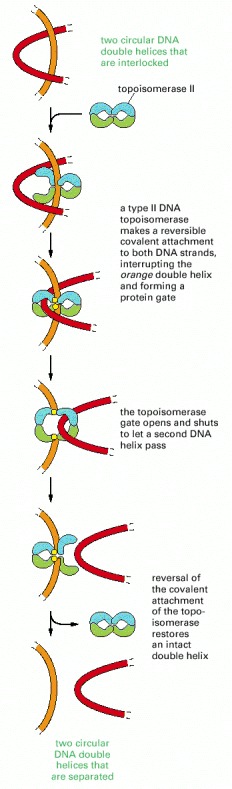
Identical reactions are used to untangle DNA inside the cell. Unlike type I topoisomerases, type II enzymes use ATP hydrolysis and some of the bacterial versions can introduce superhelical tension into DNA. Type II topoisomerases are largely confined to proliferating cells in eucaryotes; partly for that reason, they have been popular targets for anticancer drugs.
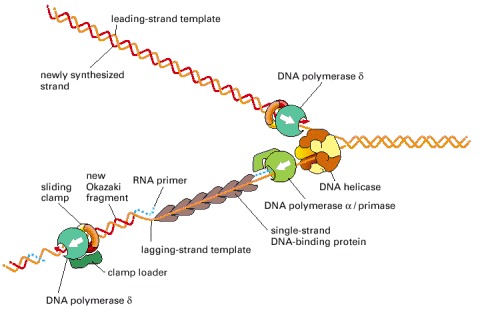
The fork is drawn to emphasize its similarity to the bacterial replication fork depicted in Figure 5-21. Although both forks use the same basic components, the mammalian fork differs in at least two important respects. First, it uses two different DNA polymerases on the lagging strand. Second, the mammalian DNA primase is a subunit of one of the lagging-strand DNA polymerases, DNA polymerase α, while that of bacteria is associated with a DNA helicase in the primosome. The polymerase α (with its associated primase) begins chains with RNA, extends them with DNA, and then hands the chains over to the second polymerase (δ), which elongates them. It is not known why eucaryotic DNA replication requires two different polymerases on the lagging strand. The major mammalian DNA helicase seems to be based on a ring formed from six different Mcm proteins; this ring may move along the leading strand, rather than along the lagging-strand template shown here.
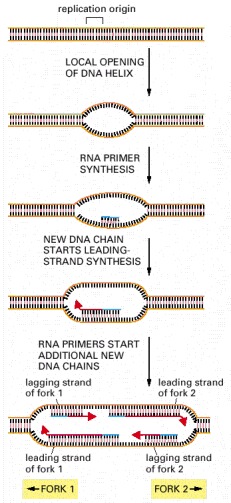
This diagram outlines the major steps involved in the initiation of replication forks at replication origins. The structure formed at the last step, in which both strands of the parental DNA helix have been separated from each other and serve as templates for DNA synthesis, is called a replication bubble.
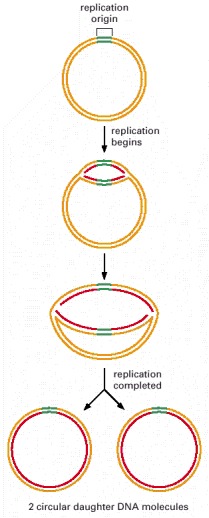
It takes E. coli about 40 minutes to duplicate its genome of 4.6 × 106 nucleotide pairs. For simplicity, no Okazaki fragments are shown on the lagging strand. What happens as the two replication forks approach each other and collide at the end of the replication cycle is not well understood, although the primosome is disassembled as part of the process.
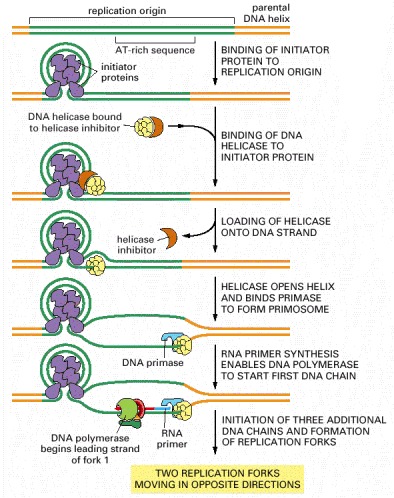
The mechanism shown was established by studies in vitro with a mixture of highly purified proteins. For E. coli DNA replication, the major initiator protein is the dnaA protein; the primosome is composed of the dnaB (DNA helicase) and dnaG (DNA primase) proteins. In solution, the helicase is bound by an inhibitor protein (the dnaC protein), which is activated by the initiator proteins to load the helicase onto DNA at the replication origin and then released. This inhibitor prevents the helicase from inappropriately entering other single-stranded stretches of DNA in the bacterial genome. Subsequent steps result in the initiation of three more DNA chains (see Figure 5-29) by a pathway whose details are incompletely specified.
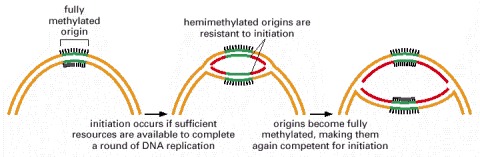
DNA methylation occurs at GATC sequences, 11 of which are found in the origin of replication (spanning about 250 nucleotide pairs). About 10 minutes after replication is initiated, the hemimethylated origins become fully methylated by a DNA methylase enzyme. As discussed earlier, the lag in methylation after the replication of GATC sequences is also used by the E. coli mismatch proofreading system to distinguish the newly synthesized DNA strand from the parental DNA strand; in that case, the relevant GATC sequences are scattered throughout the chromosome. A single enzyme, the dam methylase, is responsible for methylating E. coli GATC sequences.
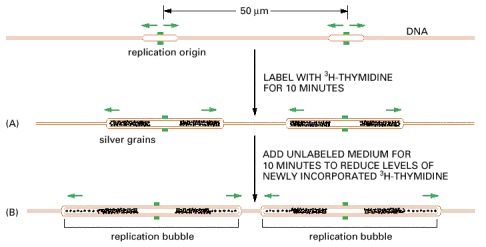
The new DNA made in human cells in culture was labeled briefly with a pulse of highly radioactive thymidine (3H-thymidine). (A) In this experiment, the cells were lysed, and the DNA was stretched out on a glass slide that was subsequently covered with a photographic emulsion. After several months the emulsion was developed, revealing a line of silver grains over the radioactive DNA. The brown DNA in this figure is shown only to help with the interpretation of the autoradiograph; the unlabeled DNA is invisible in such experiments. (B) This experiment was the same except that a further incubation in unlabeled medium allowed additional DNA, with a lower level of radioactivity, to be replicated. The pairs of dark tracks in (B) were found to have silver grains tapering off in opposite directions, demonstrating bidirectional fork movement from a central replication origin where a replication bubble forms (see Figure 5-29). A replication fork is thought to stop only when it encounters a replication fork moving in the opposite direction or when it reaches the end of the chromosome; in this way, all the DNA is eventually replicated.
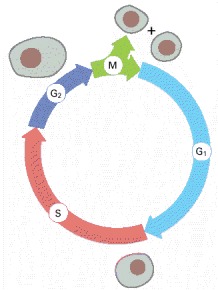
During the G1, S, and G2 phases, the cell grows continuously. During M phase growth stops, the nucleus divides, and the cell divides in two. DNA replication is confined to the part of interphase known as S phase. G1 is the gap between M phase and S phase; G2 is the gap between S phase and M phase.
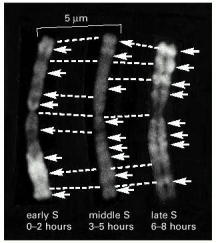
These light micrographs show stained mitotic chromosomes in which the replicating DNA has been differentially labeled during different defined intervals of the preceding S phase. In these experiments, cells were first grown in the presence of BrdU (a thymidine analog) and in the absence of thymidine to label the DNA uniformly. The cells were then briefly pulsed with thymidine in the absence of BrdU during early, middle, or late S phase. Because the DNA made during the thymidine pulse is a double helix with thymidine on one strand and BrdU on the other, it stains more darkly than the remaining DNA (which has BrdU on both strands) and shows up as a bright band (arrows) on these negatives. Broken lines connect corresponding positions on the three identical copies of the chromosome shown. (Courtesy of Elton Stubblefield.)
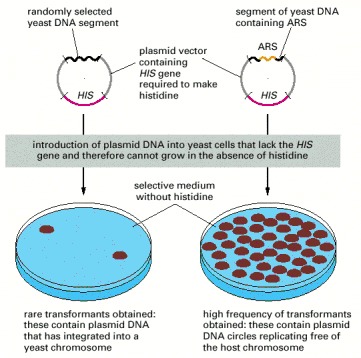
Each of the yeast DNA sequences identified in this way was called an autonomously replicating sequence (ARS), since it enables a plasmid that contains it to replicate in the host cell without having to be incorporated into a host cell chromosome.
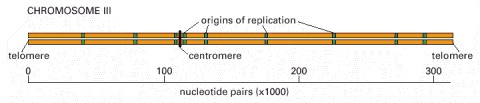
This chromosome, one of the smallest eucaryotic chromosomes known, carries a total of 180 genes. As indicated, it contains nine replication origins.
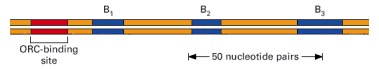
Comprising about 150 nucleotide pairs, this yeast origin (identified by the procedure shown in Figure 5-36) has a binding site for ORC, a complex of proteins that binds to every origin of replication. The origin depicted also has binding sites (B1, B2, and B3) for other required proteins, which can differ between various origins. Although best characterized in yeast, a similar ORC is used to initiate DNA replication in more complex eucaryotes, including humans.
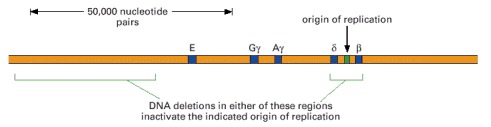
These two deletions are found separately in two individuals who suffer from thalassemia, a disorder caused by the failure to express one or more of the genes in the β-globin gene cluster shown. In both of these deletion mutants, the DNA in this region is replicated by forks that begin at replication origins outside the β-globin gene cluster. As explained in the text, the deletion on the left removes DNA sequences that control the chromatin structure of the replication origin on the right.
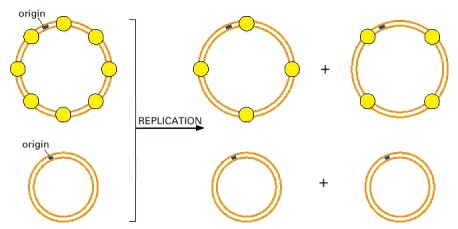
In this experiment, performed in vitro, a mixture of two different-sized circular molecules of DNA (only one of which is assembled into nucleosomes) are replicated with purified proteins. After a round of DNA replication, only the daughter DNA molecules that derived from the nucleosomal parent have inherited nucleosomes. This experiment also demonstrates that both newly synthesized DNA helices inherit old histones.
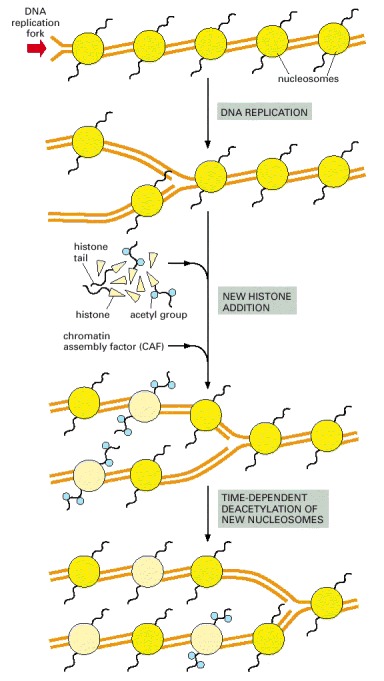
The new nucleosomes are those colored light yellow in this diagram; as indicated, some of the histones that form them initially have specifically acetylated lysine side chains (see Figure 4-35), which are later removed.
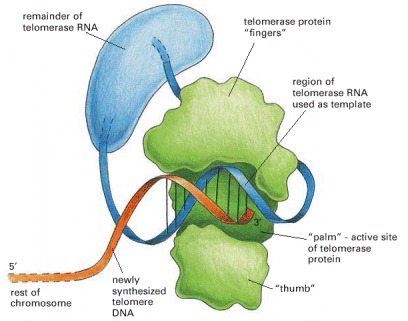
The telomerase is a protein–RNA complex that carries an RNA template for synthesizing a repeating, G-rich telomere DNA sequence. Only the part of the telomerase protein homologous to reverse transcriptase is shown here (green). A reverse transcriptase is a special form of polymerase enzyme that uses an RNA template to make a DNA strand; telomerase is unique in carrying its own RNA template with it at all times. (Modified from J. Lingner and T.R. Cech, Curr. Opin. Genet. Dev. 8:226–232, 1998.)
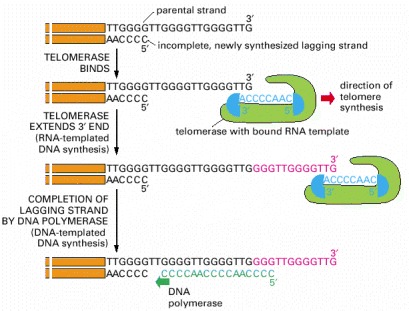
Shown here are the reactions involved in synthesizing the repeating G-rich sequences that form the ends of the chromosomes (telomeres) of diverse eucaryotic organisms. The 3′ end of the parental DNA strand is extended by RNA-templated DNA synthesis; this allows the incomplete daughter DNA strand that is paired with it to be extended in its 5′ direction. This incomplete, lagging strand is presumed to be completed by DNA polymerase α, which carries a DNA primase as one of its subunits (see Figure 5-28). The telomere sequence illustrated is that of the ciliate Tetrahymena, in which these reactions were first discovered. The telomere repeats are GGGTTG in the ciliate Tetrahymena, GGGTTA in humans, and G1–3A in the yeast S. cerevisiae.

(A) Electron micrograph of the DNA at the end of an interphase human chromosome. The chromosome was fixed, deproteinated, and artificially thickened before viewing. The loop seen here is approximately 15,000 nucleotide pairs in length. (B) Model for telomere structure. The insertion of the single-stranded end into the duplex repeats to form a t-loop is carried out and maintained by specialized proteins, schematized in green. In addition it is possible, as shown, that the chromosome end is looped once again on itself through the formation of heterochromatin adjacent to the t-loop (see Figure 4-47). (A, from J.D. Griffith et al., Cell 97:503–514, 1999. © Elsevier.)
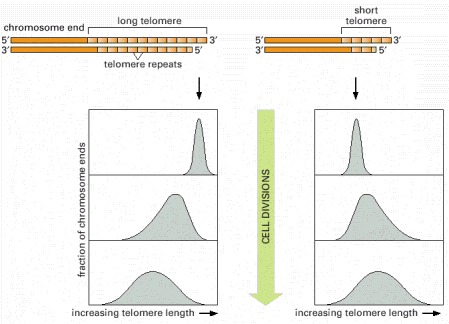
In this experiment, the telomere at one end of a particular chromosome is artificially made either longer (left) or shorter (right) than average. After many cell divisions, the chromosome recovers, showing an average telomere length and a length distribution that is typical of the other chromosomes in the yeast cell. A similar feedback mechanism for controlling telomere length has been proposed to exist for the cells in the germ-line cells of animals.
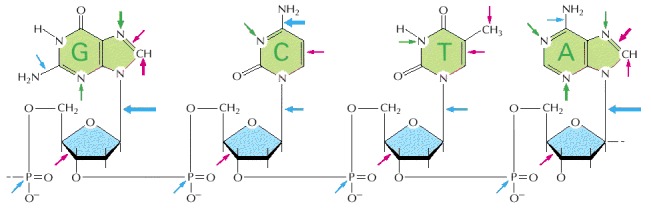
The sites on each nucleotide that are known to be modified by spontaneous oxidative damage (red arrows), hydrolytic attack (blue arrows), and uncontrolled methylation by the methyl group donor S-adenosylmethionine (green arrows) are shown, with the width of each arrow indicating the relative frequency of each event. (After T. Lindahl, Nature 362:709–715, 1993. © Macmillan Magazines Ltd.)
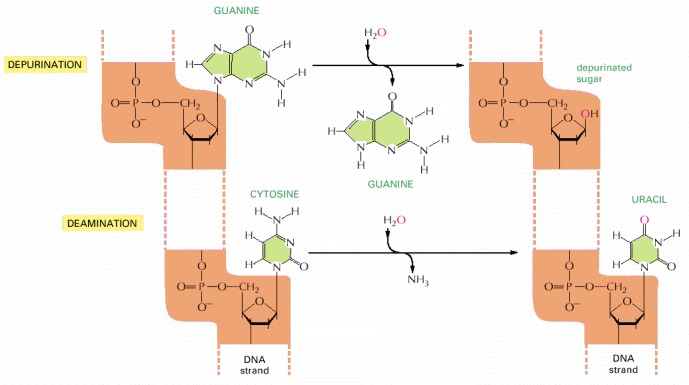
These two reactions are the most frequent spontaneous chemical reactions known to create serious DNA damage in cells. Depurination can release guanine (shown here), as well as adenine, from DNA. The major type of deamination reaction (shown here) converts cytosine to an altered DNA base, uracil, but deamination occurs on other bases as well. These reactions take place on double-helical DNA; for convenience, only one strand is shown.
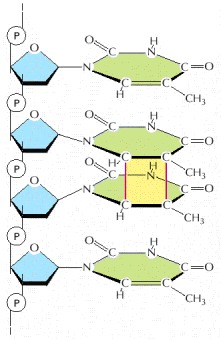
This type of damage is introduced into DNA in cells that are exposed to ultraviolet irradiation (as in sunlight). A similar dimer will form between any two neighboring pyrimidine bases (C or T residues) in DNA.
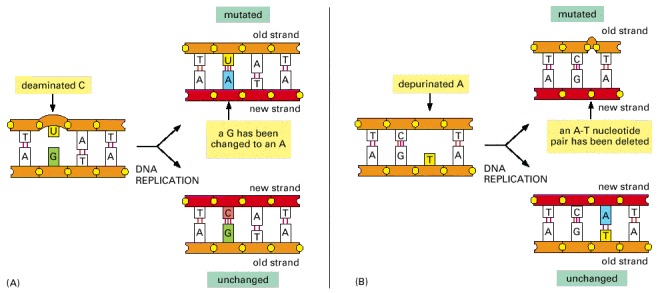
(A) Deamination of cytosine, if uncorrected, results in the substitution of one base for another when the DNA is replicated. As shown in Figure 5-47, deamination of cytosine produces uracil. Uracil differs from cytosine in its base-pairing properties and preferentially base-pairs with adenine. The DNA replication machinery therefore adds an adenine when it encounters a uracil on the template strand. (B) Depurination, if uncorrected, can lead to either the substitution or the loss of a nucleotide pair. When the replication machinery encounters a missing purine on the template strand, it may skip to the next complete nucleotide as illustrated here, thus producing a nucleotide deletion in the newly synthesized strand. Many other types of DNA damage (see Figure 5-46) also produce mutations when the DNA is replicated if left uncorrected.
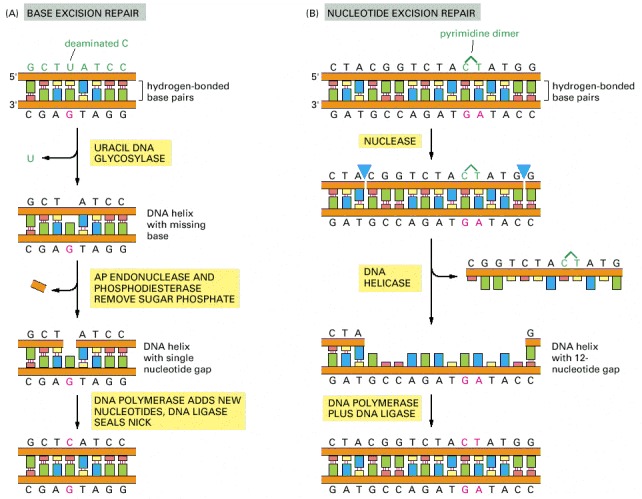
(A) Base excision repair. This pathway starts with a DNA glycosylase. Here the enzyme uracil DNA glycosylase removes an accidentally deaminated cytosine in DNA. After the action of this glycosylase (or another DNA glycosylase that recognizes a different kind of damage), the sugar phosphate with the missing base is cut out by the sequential action of AP endonuclease and a phosphodiesterase. (These same enzymes begin the repair of depurinated sites directly.) The gap of a single nucleotide is then filled by DNA polymerase and DNA ligase. The net result is that the U that was created by accidental deamination is restored to a C. The AP endonuclease derives its name from the fact that it recognizes any site in the DNA helix that contains a deoxyribose sugar with a missing base; such sites can arise either by the loss of a purine (apurinic sites) or by the loss of a pyrimidine (apyrimidinic sites). (B) Nucleotide excision repair. After a multienzyme complex has recognized a bulky lesion such as a pyrimidine dimer (see Figure 5-48), one cut is made on each side of the lesion, and an associated DNA helicase then removes the entire portion of the damaged strand. The multienzyme complex in bacteria leaves the gap of 12 nucleotides shown; the gap produced in human DNA is more than twice this size. The nucleotide excision repair machinery can recognize and repair many different types of DNA damage.
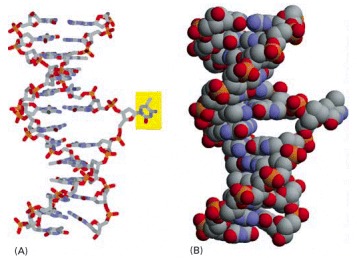
The DNA glycosylase family of enzymes recognizes specific bases in the conformation shown. Each of these enzymes cleaves the glycosyl bond that connects a particular recognized base (yellow) to the backbone sugar, removing it from the DNA. (A) Stick model; (B) space-filling model.
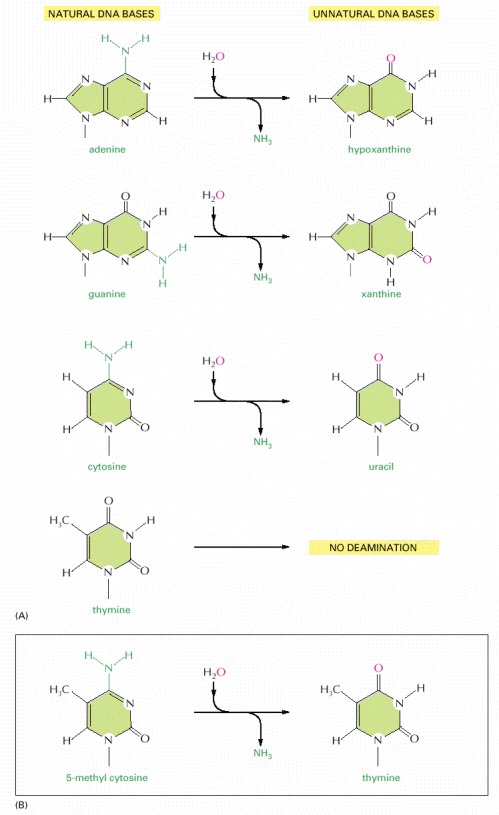
In each case the oxygen atom that is added in this reaction with water is colored red. (A) The spontaneous deamination products of A and G are recognizable as unnatural when they occur in DNA and thus are readily recognized and repaired. The deamination of C to U was previously illustrated in Figure 5-47; T has no amino group to deaminate. (B) About 3% of the C nucleotides in vertebrate DNAs are methylated to help in controlling gene expression (discussed in Chapter 7). When these 5-methyl C nucleotides are accidentally deaminated, they form the natural nucleotide T. This T would be paired with a G on the opposite strand, forming a mismatched base pair.
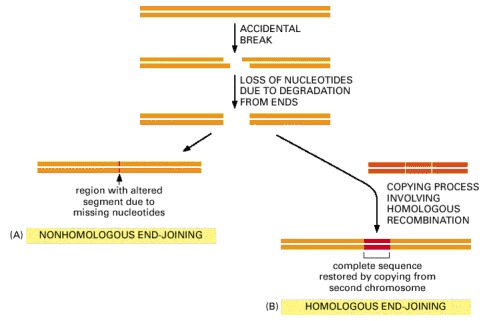
(A) Nonhomologous end-joining alters the original DNA sequence when repairing broken chromosomes. These alterations can be either deletions (as shown) or short insertions. (B) Homologous end-joining is more difficult to accomplish, but is much more precise.
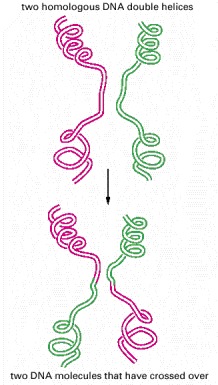
The breaking and rejoining of two homologous DNA double helices creates two DNA molecules that have “crossed over.” In meiosis, this process causes each chromosome in a germ cell to contain a mixture of maternally and paternally inherited genes.
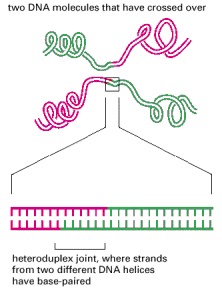
This structure unites two DNA molecules where they have crossed over. Such a joint is often thousands of nucleotides long.
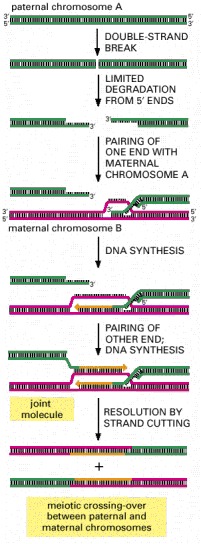
As indicated, the process begins when an endonuclease makes a double-strand break in a chromosome. An exonuclease then creates two protruding 3′ single-stranded ends, which find the homologous region of a second chromosome to begin DNA synapsis. The joint molecule formed can eventually be resolved by selective strand cuts to produce two chromosomes that have crossed over, as shown.
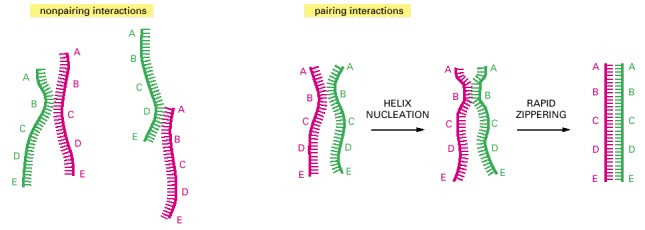
DNA double helices re-form from their separated strands in a reaction that depends on the random collision of two complementary DNA strands. The vast majority of such collisions are not productive, as shown on the left, but a few result in a short region where complementary base pairs have formed (helix nucleation). A rapid zippering then leads to the formation of a complete double helix. Through this trial-and-error process, a DNA strand will find its complementary partner even in the midst of millions of nonmatching DNA strands. A related, highly efficient trial-and-error recognition of a complementary partner DNA sequence seems to initiate all general recombination events.
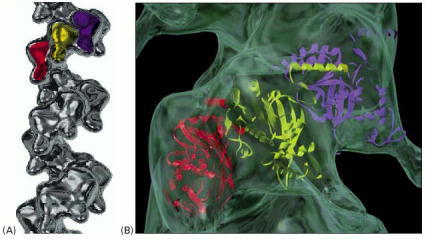
(A) The Rad51 protein bound to a DNA single strand. Rad51 is a human homolog of the bacterial RecA protein; three successive monomers in this helical filament are colored. (B) A short section of the RecA filament, with the three-dimensional structure of the protein fitted to the image of the filament determined by electron microscopy. The two DNA–protein filaments appear to be quite similar. There are about six RecA monomers per turn of the helix, holding 18 nucleotides of single-stranded DNA that is stretched out by the protein. The exact path of the DNA in this structure is not known. (A, courtesy of Edward Egelman; B, from X. Yu et al., J. Mol. Biol. 283:985–992, 1998.)
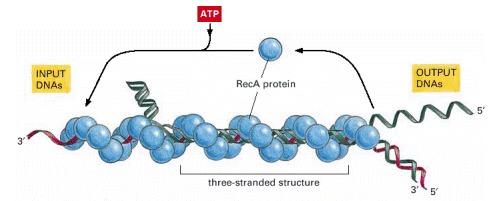
In vitro experiments show that several types of complex are formed between a DNA single strand covered with RecA protein (red) and a DNA double helix (green). First a non-base-paired complex is formed, which is converted through transient base-flipping (see Figure 5-51) to a three-stranded structure as soon as a region of homologous sequence is found. This complex is unstable because it involves an unusual form of DNA, and it spins out a DNA heteroduplex (one strand green and the other strand red) plus a displaced single strand from the original helix (green). Thus the structure shown in this diagram migrates to the left, reeling in the “input DNAs” while producing the “output DNAs.” The net result is a DNA strand exchange identical to that diagrammed earlier in Figure 5-56. (Adapted from S.C. West, Annu. Rev. Biochem. 61:603–640, 1992.)
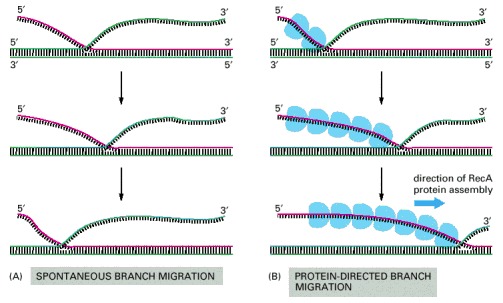
(A) Spontaneous branch migration is a back-and-forth, random-walk process, and it therefore makes little progress over long distances. (B) RecA-protein-directed branch migration proceeds at a uniform rate in one direction and may be driven by the polarized assembly of the RecA protein filament on a DNA single strand, which occurs in the direction indicated. Special DNA helicases that catalyze branch migration even more efficiently are also involved in recombination (for example see Figure 5-63).
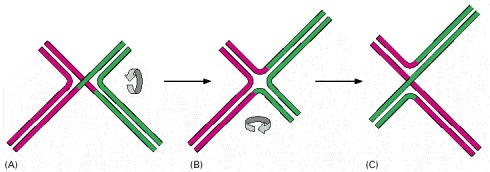
As described in the text, the synapsis step in general recombination is catalyzed by a RecA type of protein bound to a DNA single strand. This step is often followed by a reciprocal exchange of strands between two DNA double helices that have thereby paired with each other. This exchange produces a unique DNA structure known as a Holliday junction, named after the scientist who first proposed its formation. (A) The initially formed structure contains two crossing (inside) strands and two noncrossing (outside) strands. (B) An isomerization of the Holliday junction produces an open, symmetrical structure. (C) Further isomerization can interconvert the crossing and noncrossing strands, producing a structure that is otherwise the same as that in (A).
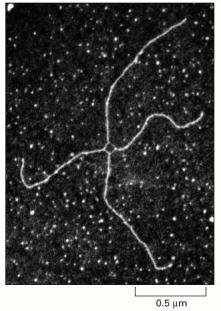
This view of the junction corresponds to the open structure illustrated in Figure 5-61B. (Courtesy of Huntington Potter and David Dressler.)
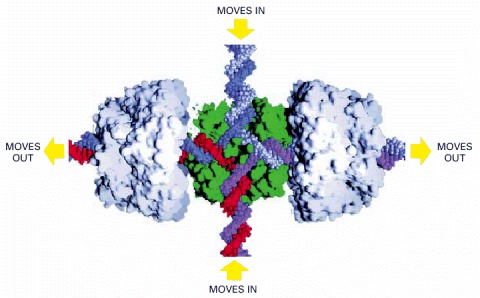
In E. coli, a tetramer of the RuvA protein (green) and two hexamers of the RuvB protein (pale gray) bind to the open form of the junction. The RuvB protein uses the energy of ATP hydrolysis to move the crossover point rapidly along the paired DNA helices, extending the heteroduplex region as shown. There is evidence that similar proteins perform this function in vertebrate cells. (Image courtesy of P. Artymiuk; modified from S.C. West, Cell 94:699–701, 1998.)
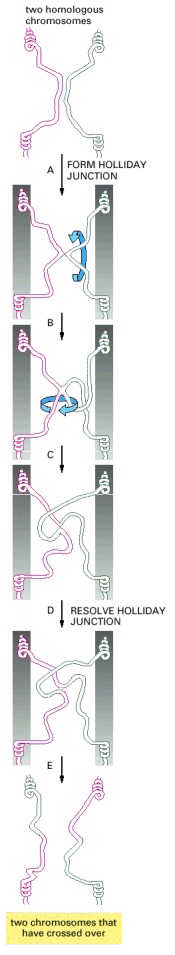
In this example, homologous regions of a red and a green chromosome have formed a Holliday junction by exchanging two strands. Cutting these two strands would terminate the exchange without crossing-over. With isomerization of the Holliday junction (steps B and C), the original noncrossing strands become the two crossing strands; cutting them now creates two DNA molecules that have crossed over (bottom). This type of isomerization may be involved in the breaking and rejoining of two homologous DNA double helices in meiotic general recombination. The grey bars in the central panels have been drawn to make it clear that the isomerization events shown can occur without disturbing the rest of the two chromosomes.
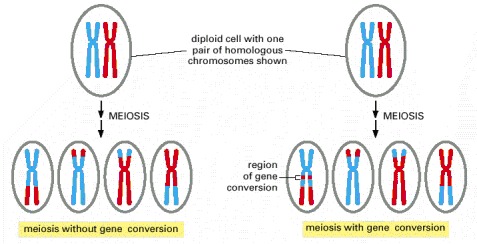
As described in Chapter 20, meiosis is the process through which a diploid cell gives rise to four haploid cells. Germ cells (eggs and sperm, for example) are produced by meiosis.
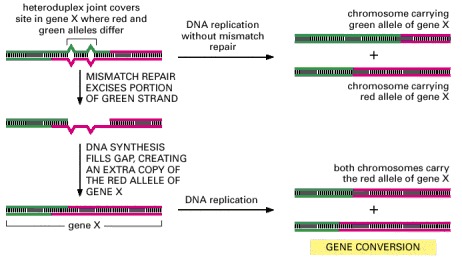
In this process, heteroduplex joints are formed at the sites of the crossing-over between homologous maternal and paternal chromosomes. If the maternal and paternal DNA sequences are slightly different, the heteroduplex joint will include some mismatched base pairs. The resulting mismatch in the double helix may then be corrected by the DNA mismatch repair machinery (see Figure 5-23), which can erase nucleotides on either the paternal or the maternal strand. The consequence of this mismatch repair is gene conversion, dectected as a deviation from the segregation of equal copies of maternal and paternal alleles that normally occurs in meiosis.
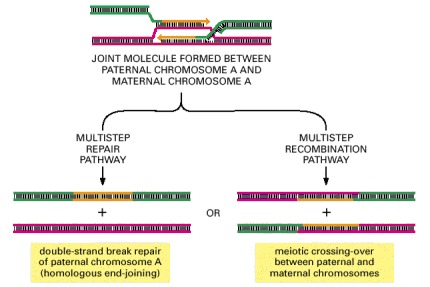
As shown previously in Figure 5-56, general recombination begins when a double-strand break is generated in one double helix (green), followed by DNA degradation and strand invasion into a homologous DNA duplex (red). New DNA synthesis (orange) follows to generate the joint molecule shown. Depending on subsequent events, resolution of the joint molecule can lead either to a precise repair of the initial double-strand break (left) or to chromosome crossing-over (right). The experimental induction of double-strand breaks at specific DNA sites has allowed the outcome of general recombination to be quantified in both mitotic and meiotic cells. More than 99% of these events fail to produce a crossover in mitotic cells, whereas crossovers are often the outcome in meiotic cells. In either case, if the maternal and paternal chromosomes differ in DNA sequence in the region of new DNA synthesis shown here, the sequence of the green DNA duplex in the region of new DNA synthesis is converted to that of the red duplex (a gene conversion).
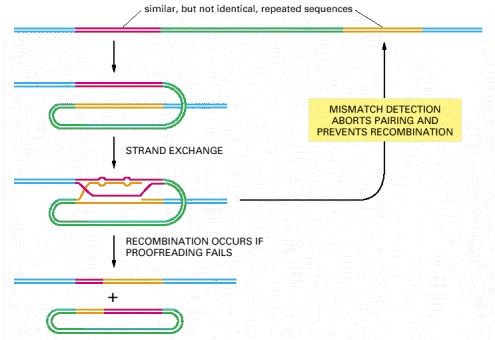
Studies with bacterial and yeast cells suggest that components of the mismatch proofreading system, diagrammed previously in Figure 5-23, have the additional function shown here.
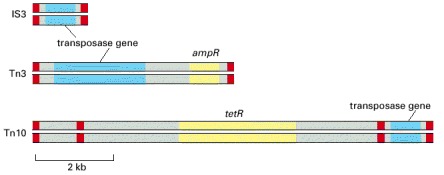
Each of these DNA elements contains a gene that encodes a transposase, an enzyme that conducts at least some of the DNA breakage and joining reactions needed for the element to move. Each mobile element also carries short DNA sequences (indicated in red) that are recognized only by the transposase encoded by that element and are necessary for movement of the element. In addition, two of the three mobile elements shown carry genes that encode enzymes that inactivate the antibiotics ampicillin (ampR) and tetracycline (tetR). The transposable element Tn10, shown in the bottom diagram, is thought to have evolved from the chance landing of two short mobile elements on either side of a tetracyclin-resistance gene; the wide use of tetracycline as an antibiotic has aided the spread of this gene through bacterial populations. The three mobile elements shown are all examples of DNA-only transposons (see text).
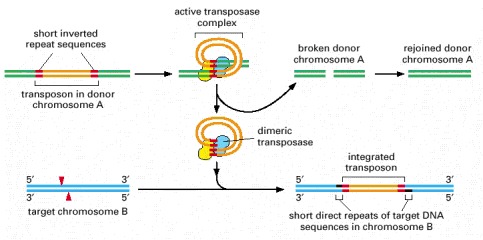
DNA-only transposons can be recognized in chromosomes by the “inverted repeat DNA sequences” (red) at their ends. Experiments show that these sequences, which can be as short as 20 nucleotides, are all that is necessary for the DNA between them to be transposed by the particular transposase enzyme associated with the element. The cut-and-paste movement of a DNA-only transposable element from one chromosomal site to another begins when the transposase brings the two inverted DNA sequences together, forming a DNA loop. Insertion into the target chromosome, catalyzed by the transposase, occurs at a random site through the creation of staggered breaks in the target chromosome (red arrowheads). As a result, the insertion site is marked by a short direct repeat of the target DNA sequence, as shown. Although the break in the donor chromosome (green) is resealed, the breakage-and-repair process often alters the DNA sequence, causing a mutation at the original site of the excised transposable element (not shown).
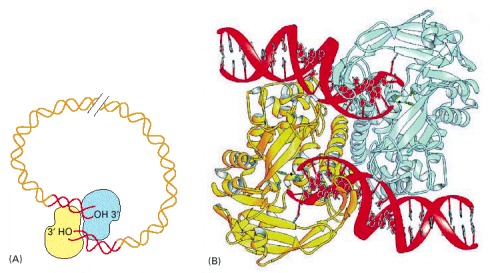
(A) Schematic view of the overall structure. (B) The detailed structure of a transposase holding the two DNA ends, whose 3′-OH groups are poised to attack a target chromosome. (B, from D.R. Davies et al., Science 289:77–85, 2000. © AAAS.)
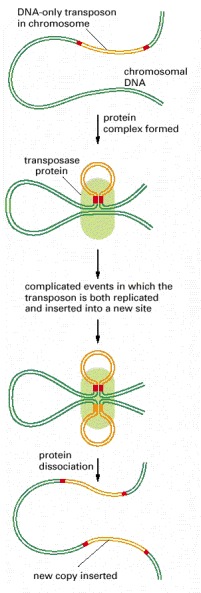
In the course of replicative transposition, the DNA sequence of the transposon is copied by DNA replication. The end products are a DNA molecule that is identical to the original donor and a target DNA molecule that has a transposon inserted into it. In general, a particular DNA-only transposon moves either by the cut-and-paste pathway shown in Figure 5-70 or by the replicative pathway outlined here. However, the two mechanisms have many enzymatic similarities, and a few transposons can move by either pathway.
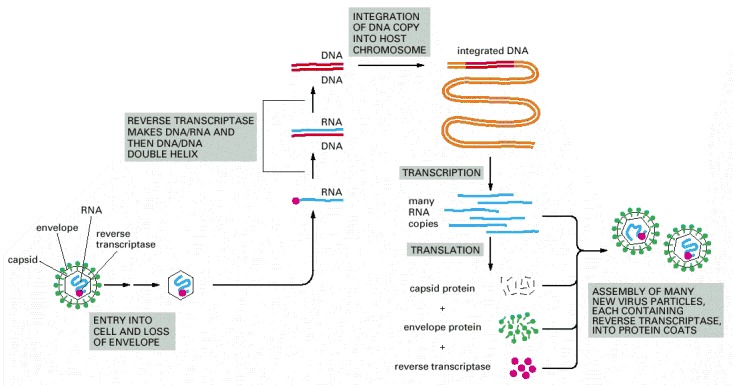
The retrovirus genome consists of an RNA molecule of about 8500 nucleotides; two such molecules are packaged into each viral particle. The enzyme reverse transcriptase first makes a DNA copy of the viral RNA molecule and then a second DNA strand, generating a double-stranded DNA copy of the RNA genome. The integration of this DNA double helix into the host chromosome is then catalyzed by a virus-encoded integrase enzyme (see Figure 5-75). This integration is required for the synthesis of new viral RNA molecules by the host cell RNA polymerase, the enzyme that transcribes DNA into RNA (discussed in Chapter 6).
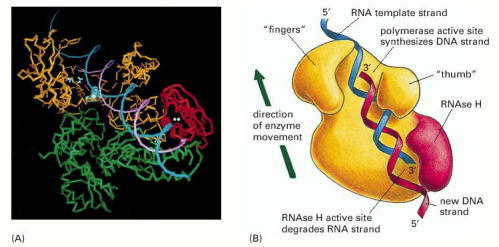
(A) The three-dimensional structure of the enzyme from HIV (the human AIDS virus) determined by x-ray crystallography. (B) A model showing the enzyme's activity on an RNA template. Note that the polymerase domain (yellow in B) has a covalently attached RNAse H (H for “Hybrid”) domain (red) that degrades an RNA strand in an RNA/DNA helix. This activity helps the polymerase to convert the initial hybrid helix into a DNA double helix (A, courtesy of Tom Steitz; B, adapted from L.A. Kohlstaedt et al., Science 256:1783–1790, 1990.)
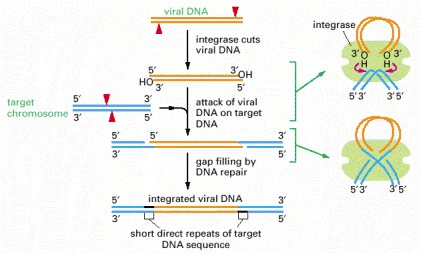
Outline of the strand-breaking and strand-rejoining events that lead to integration of the linear double-stranded DNA (orange) of a retrovirus (such as HIV) or a retroviral-like retrotransposon (such as Ty1) into the host cell chromosome (blue). In an initial step, the integrase enzyme forms a DNA loop and cuts one strand at each end of the viral DNA sequence, exposing a protruding 3′-OH group. Each of these 3′-OH ends then directly attacks a phosphodiester bond on opposite strands of a randomly selected site on a target chromosome (red arrowheads). This inserts the viral DNA sequence into the target chromosome, leaving short gaps on each side that are filled in by DNA repair processes. Because of the gap filling, this type of mechanism (like that of cut-and-paste transposons) leaves short repeats of target DNA sequence (black) on each side of the integrated DNA segment; these are 3–12 nucleotides long, depending on the integrase enzyme.
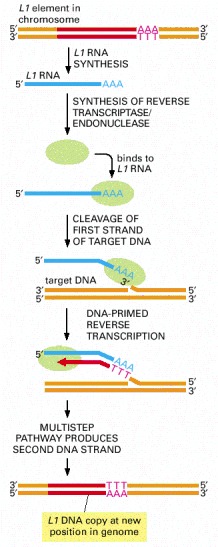
Transposition by the L1 element (red) begins when an endonuclease attached to the L1 reverse transcriptase and the L1 RNA (blue) makes a nick in the target DNA at the point at which insertion will occur. This cleavage releases a 3′-OH DNA end in the target DNA, which is then used as a primer for the reverse transcription step shown. This generates a single-stranded DNA copy of the element that is directly linked to the target DNA. In subsequent reactions, not yet understood in detail, further processing of the single-stranded DNA copy results in the generation of a new double-stranded DNA copy of the L1 element that is inserted at the site where the initial nick was made.
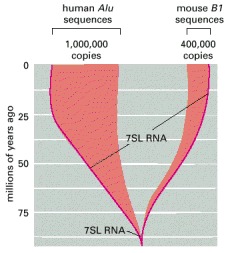
Both of these transposable DNA sequences are thought to have evolved from the essential 7SL RNA gene which encodes the SRP RNA (see Figure 12-41). On the basis of the species distribution and sequence similarity of these highly repeated elements, the major expansion in copy numbers seems to have occurred independently in mice and humans (see Figure 5-78). (Adapted from P.L. Deininger and G.R. Daniels, Trends Genet. 2:76–80, 1986 and International Human Genome Sequencing Consortium, Nature 409:860–921, 2001.)
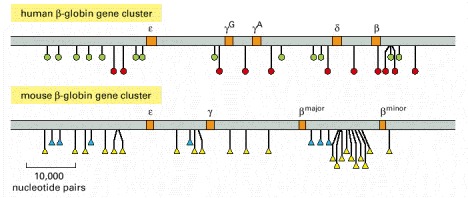
This stretch of human genome contains five functional β-globin-like genes (orange); the comparable region from the mouse genome has only four. The positions of the human Alu sequence are indicated by green circles, and the human L1 sequences by red circles. The mouse genome contains different but related transposable elements: the positions of B1 elements (which are related to the human Alu sequences) are indicated by blue triangles, and the positions of the mouse L1 elements (which are related to the human L1 sequences) are indicated by yellow triangles. Because the DNA sequences and positions of the transposable elements found in the mouse and human β-globin gene clusters are so different, it is believed that they accumulated in each of these genomes independently, relatively recently in evolutionary time. (Courtesy of Ross Hardison and Webb Miller.)
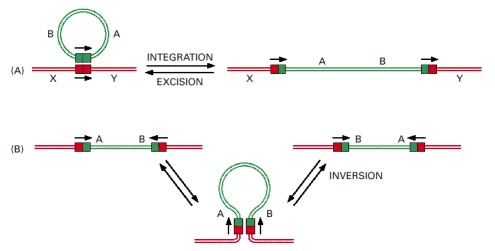
The only difference between the reactions in (A) and B) is the relative orientation of the two DNA sites (indicated by arrows) at which a site-specific recombination event occurs. (A) Through an integration reaction, a circular DNA molecule can become incorporated into a second DNA molecule; by the reverse reaction (excision), it can exit to reform the original DNA circle. Bacteriophage lambda and other bacterial viruses move in and out of their host chromosomes in precisely this way. (B) Conservative site-specific recombination can also invert a specific segment of DNA in a chromosome. A well-studied example of DNA inversion through site-specific recombination occurs in the bacterium Salmonella typhimurium, an organism that is a major cause of food poisoning in humans; the inversion of a DNA segment changes the type of flagellum that is produced by the bacterium (see Figure 7-64).
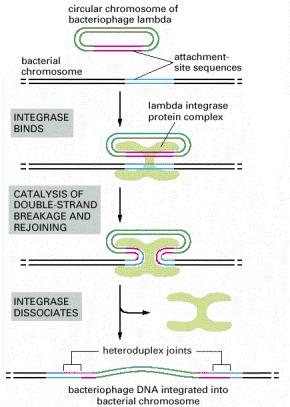
In this example of site-specific recombination, the lambda integrase enzyme binds to a specific “attachment site” DNA sequence on each chromosome, where it makes cuts that bracket a short homologous DNA sequence. The integrase then switches the partner strands and reseals them to form a heteroduplex joint that is seven nucleotide pairs long. A total of four strand-breaking and strand-joining reactions is required; for each of them, the energy of the cleaved phosphodiester bond is stored in a transient covalent linkage between the DNA and the enzyme, so that DNA strand resealing occurs without a requirement for ATP or DNA ligase.
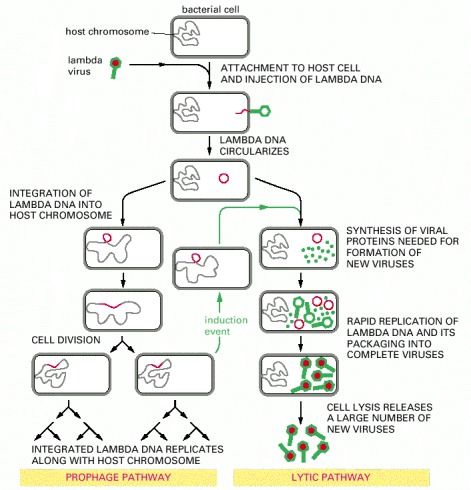
The double-stranded DNA lambda genome contains 50,000 nucleotide pairs and encodes 50–60 different proteins. When the lambda DNA enters the cell, the ends join to form a circular DNA molecule. This bacteriophage can multiply in E. coli by a lytic pathway, which destroys the cell, or it can enter a latent prophage state. Damage to a cell carrying a lambda prophage induces the prophage to exit from the host chromosome and shift to lytic growth (green arrows). Both the entrance of the lambda DNA to, and its exit from, the bacterial chromosome are accomplished by a conservative site-specific recombination event, catalyzed by the lambda integrase enzyme (see Figure 5-80).
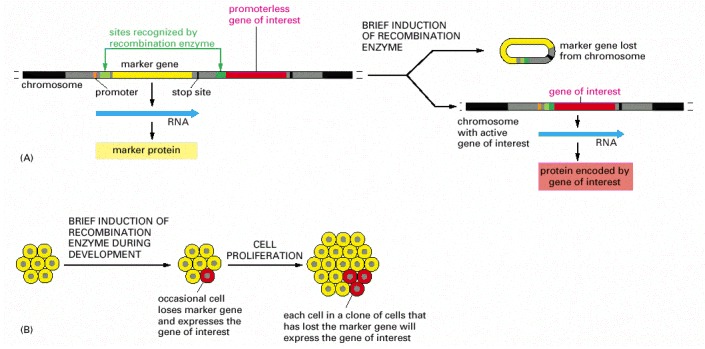
This technique requires the insertion of two specially engineered DNA molecules into the animal's germ line. (A) The DNA molecule shown has been engineered with specific recognition sites (green) so that the gene of interest (red) is transcribed only after a site-specific recombination enzyme that uses these sites is induced. As shown on the right, this induction removes a marker gene (yellow) and brings the promoter DNA (orange) adjacent to the gene of interest. The recombination enzyme is inducible, because it is encoded by a second DNA molecule (not shown) that has been engineered to ensure that the enzyme is made only when the animal is treated with a special small molecule or its temperature is raised. (B) Transient induction of the recombination enzyme causes a brief burst of synthesis of that enzyme, which in turn causes a DNA rearrangement in an occasional cell. For this cell and all its progeny, the marker gene is inactivated and the gene of interest is simultaneously activated (as shown in A). Those clones of cells in the developing animal that express the gene of interest can be identified by their loss of the marker protein. This technique is widely used in mice and Drosophila, because it allows one to study the effect of expressing any gene of interest in a group of cells in an intact animal. In one version of the technique, the Cre recombination enzyme of bacteriophage P1 is employed along with its loxP recognition sites (see pp. 542–543).