Chapter 3 - Figures
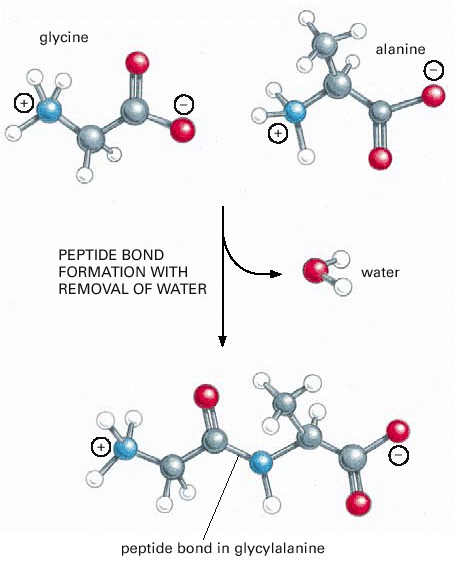
This covalent bond forms when the carbon atom from the carboxyl group of one amino acid shares electrons with the nitrogen atom (blue) from the amino group of a second amino acid. As indicated, a molecule of water is lost in this condensation reaction.
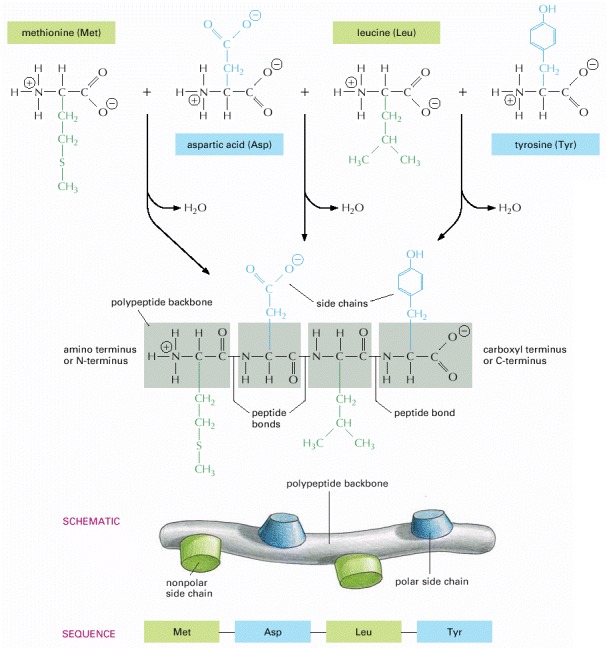
A protein consists of a polypeptide backbone with attached side chains. Each type of protein differs in its sequence and number of amino acids; therefore, it is the sequence of the chemically different side chains that makes each protein distinct. The two ends of a polypeptide chain are chemically different: the end carrying the free amino group (NH3 +, also written NH2) is the amino terminus, or N-terminus, and that carrying the free carboxyl group (COO–, also written COOH) is the carboxyl terminus or C-terminus. The amino acid sequence of a protein is always presented in the N-to-C direction, reading from left to right.
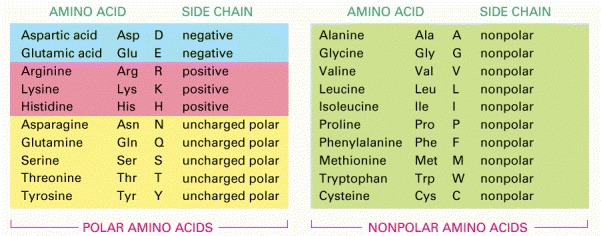
Both three-letter and one-letter abbreviations are listed. As shown, there are equal numbers of polar and nonpolar side chains. For their atomic structures, see Panel 3-1 (pp. 132–133).
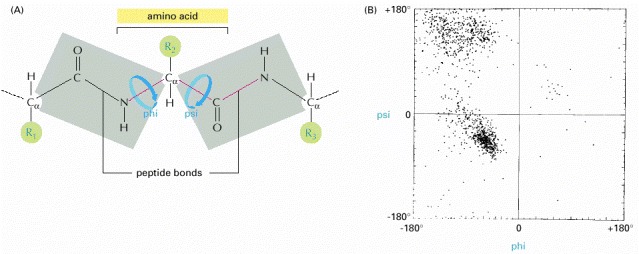
(A) Each amino acid contributes three bonds (red) to the backbone of the chain. The peptide bond is planar (gray shading) and does not permit rotation. By contrast, rotation can occur about the Cα–C bond, whose angle of rotation is called psi (ψ), and about the N–Cα bond, whose angle of rotation is called phi (ϕ). By convention, an R group is often used to denote an amino acid side chain (green circles). (B) The conformation of the main-chain atoms in a protein is determined by one pair of ϕ and ψ angles for each amino acid; because of steric collisions between atoms within each amino acid, most pairs of ϕ and ψ angles do not occur. In this so-called Ramachandran plot, each dot represents an observed pair of angles in a protein. (B, from J. Richardson, Adv. Prot. Chem. 34:174–175, 1981. © Academic Press.)
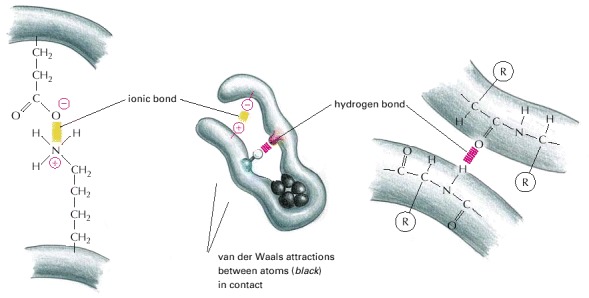
Although a single one of these bonds is quite weak, many of them often form together to create a strong bonding arrangement, as in the example shown. As in the previous figure, R is used as a general designation for an amino acid side chain.
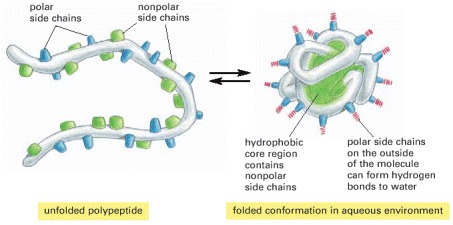
The polar amino acid side chains tend to gather on the outside of the protein, where they can interact with water; the nonpolar amino acid side chains are buried on the inside to form a tightly packed hydrophobic core of atoms that are hidden from water. In this schematic drawing, the protein contains only about 30 amino acids.
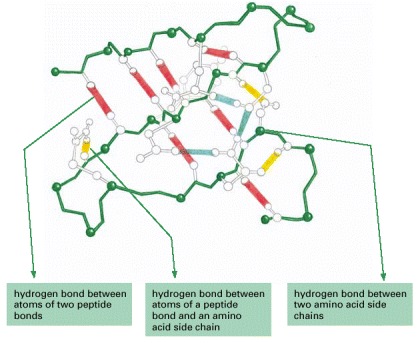
Large numbers of hydrogen bonds form between adjacent regions of the folded polypeptide chain and help stabilize its three-dimensional shape. The protein depicted is a portion of the enzyme lysozyme, and the hydrogen bonds between the three possible pairs of partners have been differently colored, as indicated. (After C.K. Mathews and K.E. van Holde, Biochemistry. Redwood City, CA: Benjamin/Cummings, 1996.)
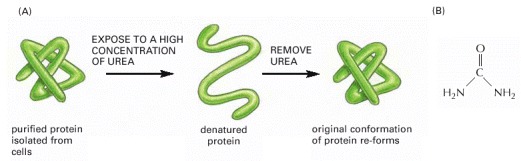
(A) This experiment demonstrates that the conformation of a protein is determined solely by its amino acid sequence. (B) The structure of urea. Urea is very soluble in water and unfolds proteins at high concentrations, where there is about one urea molecule for every six water molecules.
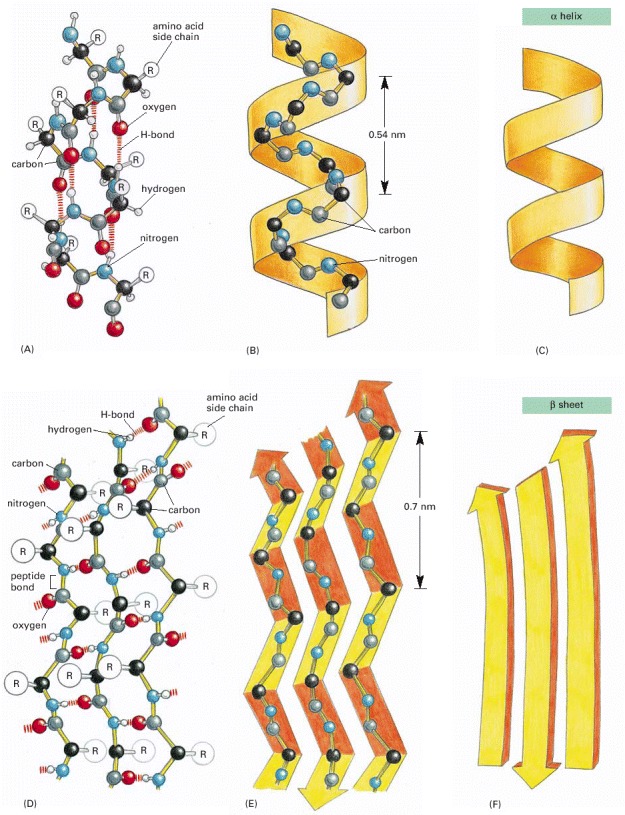
(A, B, and C) The α helix. The N–H of every peptide bond is hydrogen-bonded to the C=O of a neighboring peptide bond located four peptide bonds away in the same chain. (D, E, and F) The β sheet. In this example, adjacent peptide chains run in opposite (antiparallel) directions. The individual polypeptide chains (strands) in a β sheet are held together by hydrogen-bonding between peptide bonds in different strands, and the amino acid side chains in each strand alternately project above and below the plane of the sheet. (A) and (D) show all the atoms in the polypeptide backbone, but the amino acid side chains are truncated and denoted by R. In contrast, (B) and (E) show the backbone atoms only, while (C) and (F) display the shorthand symbols that are used to represent the α helix and the β sheet in ribbon drawings of proteins (see Panel 3-2B).
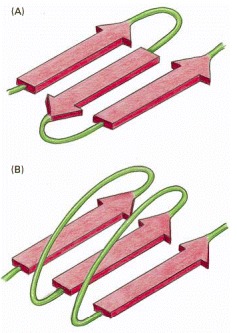
(A) An antiparallel β sheet (see Figure 3-9D). (B) A parallel β sheet. Both of these structures are common in proteins.
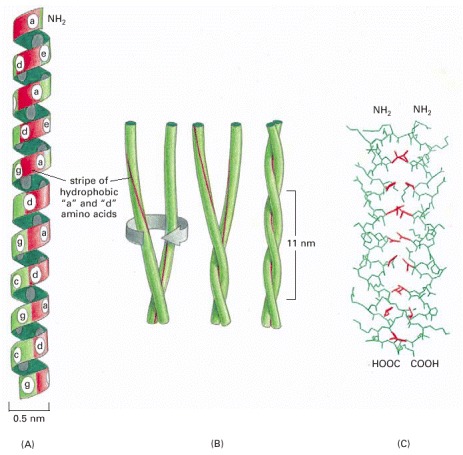
(A) A single α helix, with successive amino acid side chains labeled in a sevenfold sequence, “abcdefg” (from bottom to top). Amino acids “a” and “d” in such a sequence lie close together on the cylinder surface, forming a “stripe” (red) that winds slowly around the α helix. Proteins that form coiled-coils typically have nonpolar amino acids at positions “a” and “d.” Consequently, as shown in (B), the two α helices can wrap around each other with the nonpolar side chains of one α helix interacting with the nonpolar side chains of the other, while the more hydrophilic amino acid side chains are left exposed to the aqueous environment. (C) The atomic structure of a coiled-coil determined by x-ray crystallography. The red side chains are nonpolar.
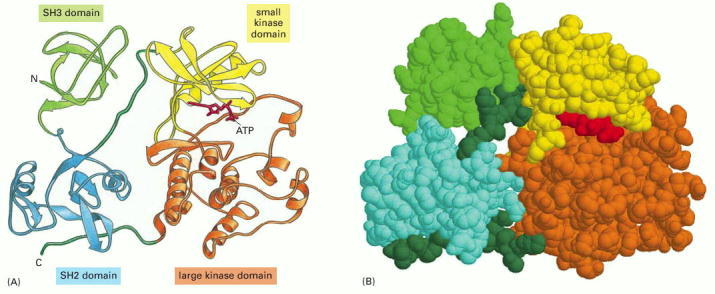
In the Src protein shown, two of the domains form a protein kinase enzyme, while the SH2 and SH3 domains perform regulatory functions. (A) A ribbon model, with ATP substrate in red. (B) A spacing-filling model, with ATP substrate in red. Note that the site that binds ATP is positioned at the interface of the two domains that form the kinase. The detailed structure of the SH2 domain is illustrated in Panel 3-2 (pp. 138–139).
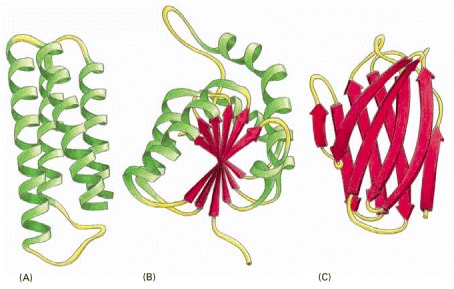
(A) Cytochrome b562, a single-domain protein involved in electron transport in mitochondria. This protein is composed almost entirely of α helices. (B) The NAD-binding domain of the enzyme lactic dehydrogenase, which is composed of a mixture of α helices and β sheets. (C) The variable domain of an immunoglobulin (antibody) light chain, composed of a sandwich of two β sheets. In these examples, the α helices are shown in green, while strands organized as β sheets are denoted by red arrows.
Note that the polypeptide chain generally traverses back and forth across the entire domain, making sharp turns only at the protein surface. It is the protruding loop regions (yellow) that often form the binding sites for other molecules. (Adapted from drawings courtesy of Jane Richardson.)
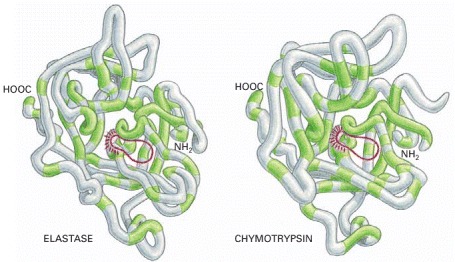
The backbone conformations of elastase and chymotrypsin. Although only those amino acids in the polypeptide chain shaded in green are the same in the two proteins, the two conformations are very similar nearly everywhere. The active site of each enzyme is circled in red; this is where the peptide bonds of the proteins that serve as substrates are bound and cleaved by hydrolysis. The serine proteases derive their name from the amino acid serine, whose side chain is part of the active site of each enzyme and directly participates in the cleavage reaction.
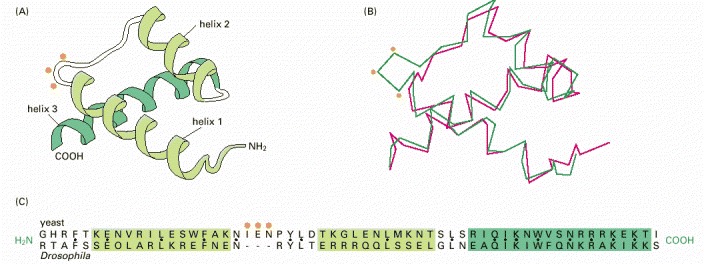
(A) A ribbon model of the structure common to both proteins. (B) A trace of the α-carbon positions. The three-dimensional structures shown were determined by x-ray crystallography for the yeast α2 protein (green) and the Drosophila engrailed protein (red). (C) A comparison of amino acid sequences for the region of the proteins shown in (A) and (B). Black dots mark sites with identical amino acids. Orange dots indicate the position of a three amino acid insert in the α2 protein. (Adapted from C. Wolberger et al., Cell 67:517–528, 1991.)
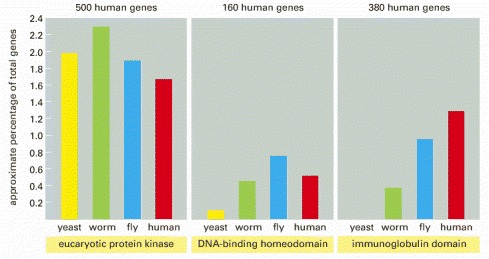
Note that one of the three domains selected, the immunoglobulin domain, has been a relatively late addition, and its relative abundance has increased in the vertebrate lineage. The estimates of human gene numbers are approximate.
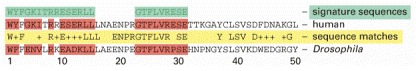
The two short sequences of 15 and 9 amino acids shown (green) can be used to search large databases for a protein domain that is found in many proteins, the SH2 domain. Here, the first 50 amino acids of the SH2 domain of 100 amino acids is compared for the human and Drosophila Src protein (see Figure 3-12). In the computer-generated sequence comparison (yellow row), exact matches between the human and Drosophila proteins are noted by the one-letter abbreviation for the amino acid; the positions with a similar but nonidentical amino acid are denoted by +, and nonmatches are blank. In this diagram, wherever one or both proteins contain an exact match to a position in the green sequences, both aligned sequences are colored red.
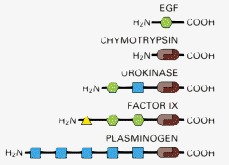
An extensive shuffling of blocks of protein sequence (protein domains) has occurred during protein evolution. Those portions of a protein denoted by the same shape and color in this diagram are evolutionarily related. Serine proteases like chymotrypsin are formed from two domains (brown). In the three other proteases shown, which are highly regulated and more specialized, these two protease domains are connected to one or more domains homologous to domains found in epidermal growth factor (EGF; green), to a calcium-binding protein (yellow), or to a “kringle” domain (blue) that contains three internal disulfide bridges. Chymotrypsin is illustrated in Figure 3-14.
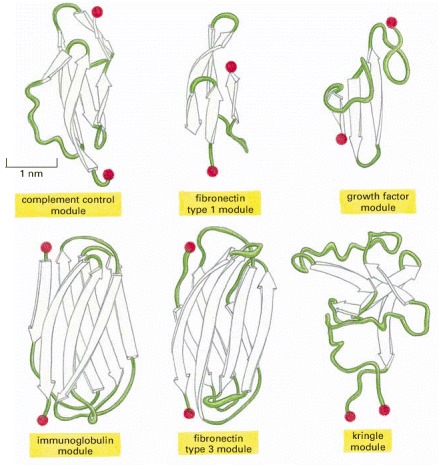
In these ribbon diagrams, β-sheet strands are shown as arrows, and the N- and C-termini are indicated by red spheres. (Adapted from M. Baron, D.G. Norman, and I.D. Campbell, Trends Biochem. Sci. 16:13–17, 1991, and D.J. Leahy et al., Science 258:987–991, 1992.)
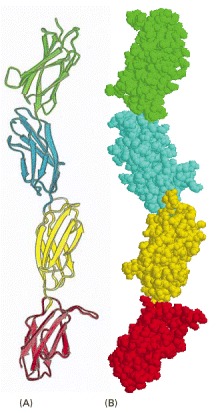
Four fibronectin type 3 modules (see Figure 3-19) from the extracellular matrix molecule fibronectin are illustrated in (A) ribbon and (B) space-filling models. (Adapted from D.J. Leahy, I. Aukhil, and H.P. Erickson, Cell 84:155–164, 1996.)
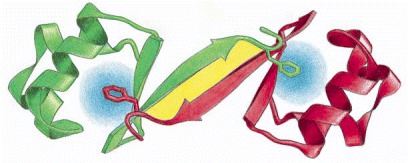
The Cro repressor protein from bacteriophage lambda binds to DNA to turn off viral genes. Its two identical subunits bind head-to-head, held together by a combination of hydrophobic forces (blue) and a set of hydrogen bonds (yellow region). (Adapted from D.H. Ohlendorf, D.E. Tronrud, and B.W. Matthews, J. Mol. Biol. 280:129–136, 1998.)
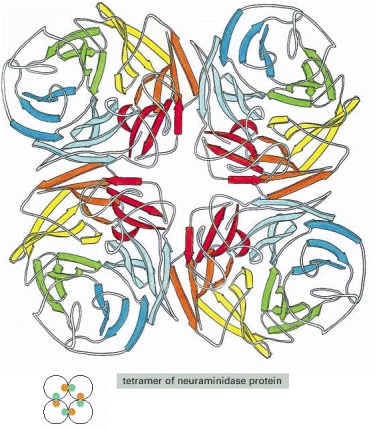
The enzyme neuraminidase exists as a ring of four identical polypeptide chains. The small diagram shows how the repeated use of the same binding interaction forms the structure.
Hemoglobin is an abundant protein in red blood cells that contains two copies of α globin and two copies of β globin. Each of these four polypeptide chains contains a heme molecule (red), which is the site where oxygen (O2) is bound. Thus, each molecule of hemoglobin in the blood carries four molecules of oxygen.
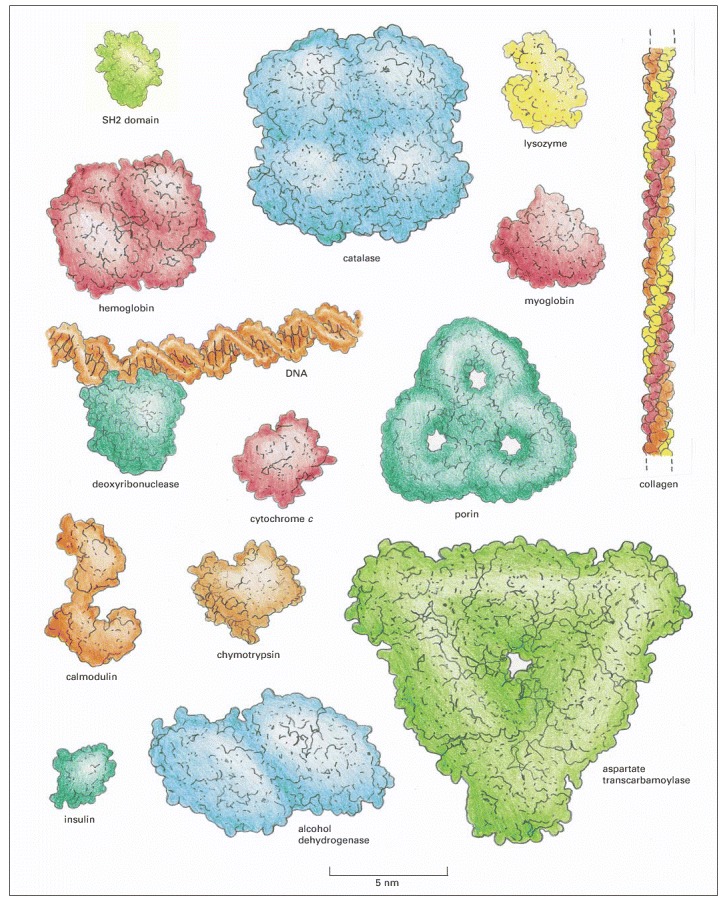
For comparison, a DNA molecule bound to a protein is also illustrated. These space-filling models represent a range of sizes and shapes. Hemoglobin, catalase, porin, alcohol dehydrogenase, and aspartate transcarbamoylase are formed from multiple copies of subunits. The SH2 domain (top left) is presented in detail in Panel 3-2 (pp. 138–139). (After David S. Goodsell, Our Molecular Nature. New York: Springer-Verlag, 1996.)
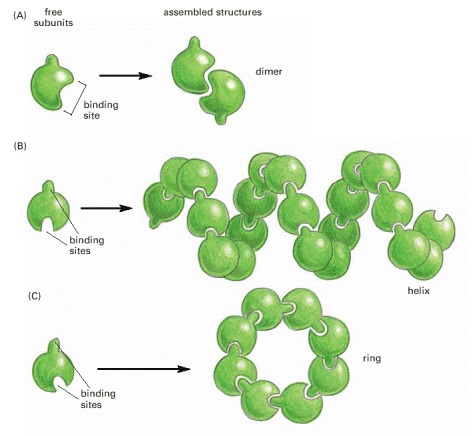
(A) A protein with just one binding site can form a dimer with another identical protein. (B) Identical proteins with two different binding sites often form a long helical filament. (C) If the two binding sites are disposed appropriately in relation to each other, the protein subunits may form a closed ring instead of a helix. (For an example of A, see Figure 3-21; for an example of C, see Figure 3-22.)
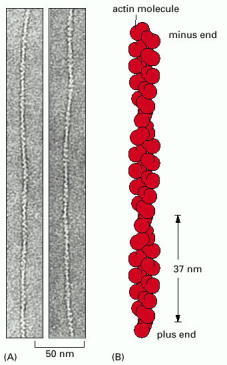
(A) Transmission electron micrographs of negatively stained actin filaments. (B) The helical arrangement of actin molecules in an actin filament. (A, courtesy of Roger Craig.)
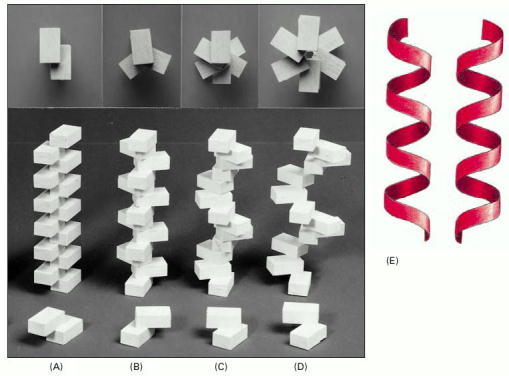
(A–D) A helix forms when a series of subunits bind to each other in a regular way. At the bottom, the interaction between two subunits is shown; behind them are the helices that result. These helices have two (A), three (B), and six (C and D) subunits per helical turn. At the top, the arrangement of subunits has been photographed from directly above the helix. Note that the helix in (D) has a wider path than that in (C), but the same number of subunits per turn. (E) A helix can be either right-handed or left-handed. As a reference, it is useful to remember that standard metal screws, which insert when turned clockwise, are right-handed. Note that a helix retains the same handedness when it is turned upside down.
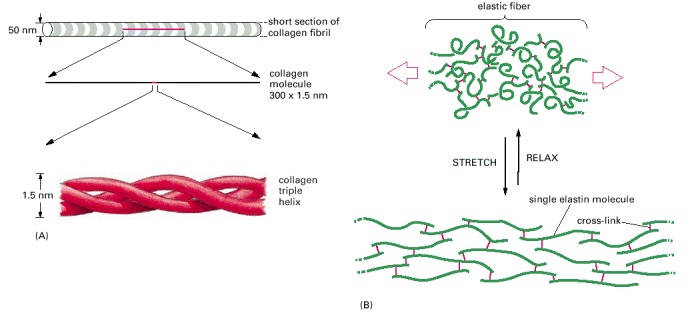
(A) Collagen is a triple helix formed by three extended protein chains that wrap around one another (bottom). Many rodlike collagen molecules are cross-linked together in the extracellular space to form unextendable collagen fibrils (top) that have the tensile strength of steel. The striping on the collagen fibril is caused by the regular repeating arrangement of the collagen molecules within the fibril. (B) Elastin polypeptide chains are cross-linked together to form rubberlike, elastic fibers. Each elastin molecule uncoils into a more extended conformation when the fiber is stretched and recoils spontaneously as soon as the stretching force is relaxed.
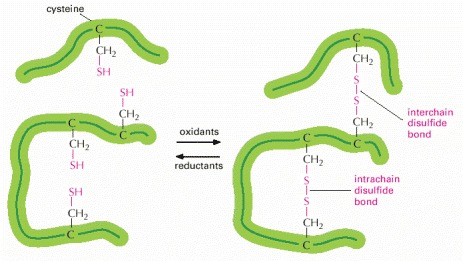
This diagram illustrates how covalent disulfide bonds form between adjacent cysteine side chains. As indicated, these cross-linkages can join either two parts of the same polypeptide chain or two different polypeptide chains. Since the energy required to break one covalent bond is much larger than the energy required to break even a whole set of noncovalent bonds (see Table 2-2, p. 57), a disulfide bond can have a major stabilizing effect on a protein.
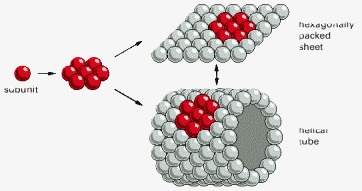
Hexagonally packed globular protein subunits can form either a flat sheet or a tube.
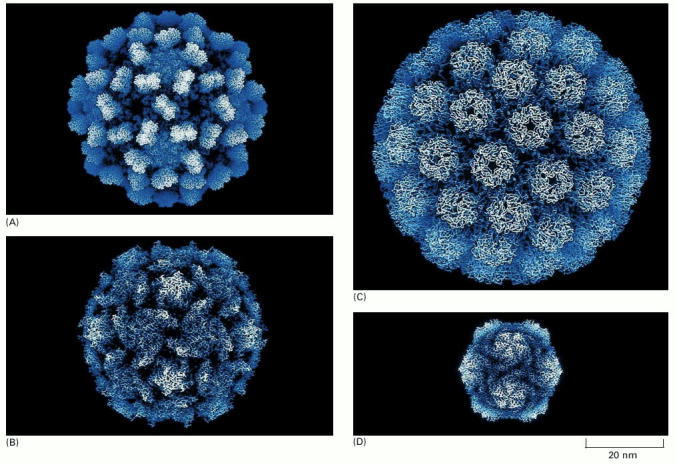
(A) Tomato bushy stunt virus; (B) poliovirus; (C) simian virus 40 (SV40); (D) satellite tobacco necrosis virus. The structures of all of these capsids have been determined by x-ray crystallography and are known in atomic detail. (Courtesy of Robert Grant, Stephan Crainic, and James M. Hogle.)
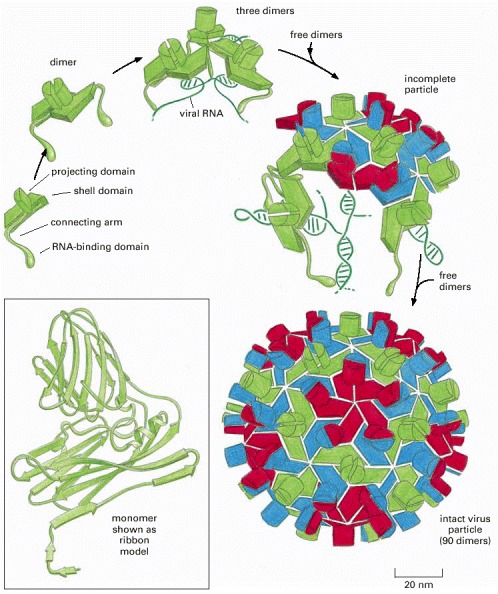
In many viruses, identical protein subunits pack together to create a spherical shell (a capsid) that encloses the viral genome, composed of either RNA or DNA (see also Figure 3-31). For geometric reasons, no more than 60 identical subunits can pack together in a precisely symmetric way. If slight irregularities are allowed, however, more subunits can be used to produce a larger capsid. The tomato bushy stunt virus (TBSV) shown here, for example, is a spherical virus about 33 nm in diameter formed from 180 identical copies of a 386 amino acid capsid protein plus an RNA genome of 4500 nucleotides. To construct such a large capsid, the protein must be able to fit into three somewhat different environments, each of which is differently colored in the virus particle shown here. The postulated pathway of assembly is shown; the precise three-dimensional structure has been determined by x-ray diffraction. (Courtesy of Steve Harrison.)

(A) An electron micrograph of the viral particle, which consists of a single long RNA molecule enclosed in a cylindrical protein coat composed of identical protein subunits. (B) A model showing part of the structure of TMV. A single-stranded RNA molecule of 6000 nucleotides is packaged in a helical coat constructed from 2130 copies of a coat protein 158 amino acids long. Fully infective viral particles can self-assemble in a test tube from purified RNA and protein molecules. (A, courtesy of Robley Williams; B, courtesy of Richard J. Feldmann.)
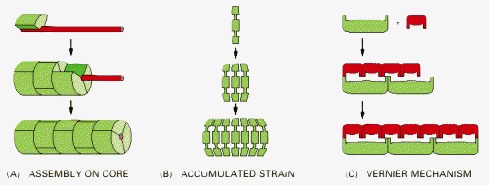
(A) Coassembly along an elongated core protein or other macromolecule that acts as a measuring device. (B) Termination of assembly because of strain that accumulates in the polymeric structure as additional subunits are added, so that beyond a certain length the energy required to fit another subunit onto the chain becomes excessively large. (C) A vernier type of assembly, in which two sets of rodlike molecules differing in length form a staggered complex that grows until their ends exactly match. The name derives from a measuring device based on the same principle, used in mechanical instruments.
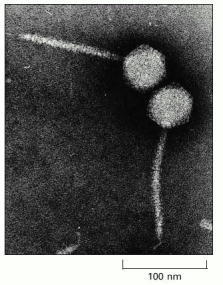
The tip of the virus tail attaches to a specific protein on the surface of a bacterial cell, after which the tightly packaged DNA in the head is injected through the tail into the cell. The tail has a precise length, determined by the mechanism shown in Figure 3-34A.
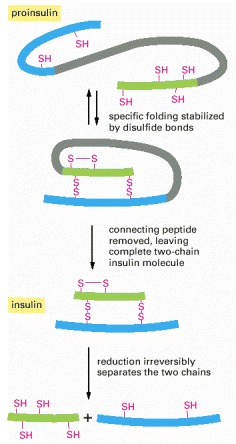
The polypeptide hormone insulin cannot spontaneously re-form efficiently if its disulfide bonds are disrupted. It is synthesized as a larger protein (proinsulin) that is cleaved by a proteolytic enzyme after the protein chain has folded into a specific shape. Excision of part of the proinsulin polypeptide chain removes some of the information needed for the protein to fold spontaneously into its normal conformation once it has been denatured and its two polypeptide chains separated.
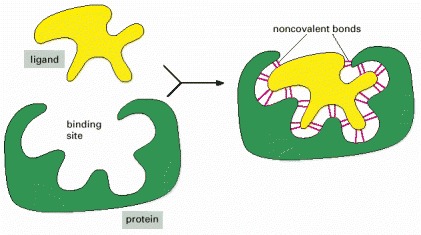
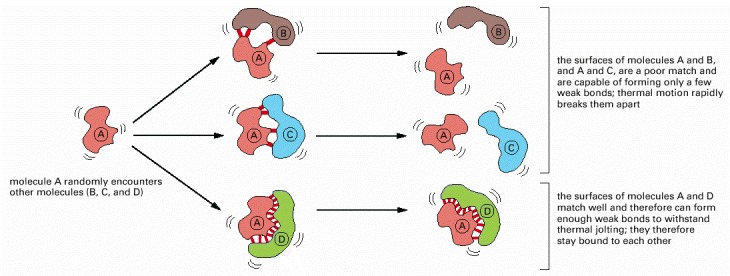
Many weak bonds are needed to enable a protein to bind tightly to a second molecule, which is called a ligand for the protein. A ligand must therefore fit precisely into a protein’s binding site, like a hand into a glove, so that a large number of noncovalent bonds can be formed between the protein and the ligand.
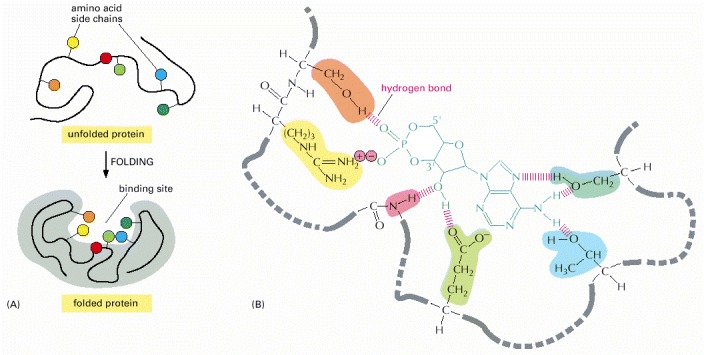
(A) The folding of the polypeptide chain typically creates a crevice or cavity on the protein surface. This crevice contains a set of amino acid side chains disposed in such a way that they can make noncovalent bonds only with certain ligands. (B) A close-up of an actual binding site showing the hydrogen bonds and ionic interactions formed between a protein and its ligand (in this example, cyclic AMP is the bound ligand).
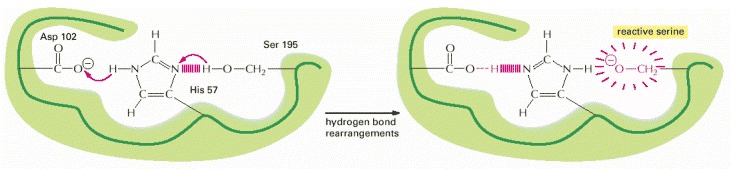
This example is the “catalytic triad” found in chymotrypsin, elastase, and other serine proteases (see Figure 3-14). The aspartic acid side chain (Asp 102) induces the histidine (His 57) to remove the proton from serine 195. This activates the serine to form a covalent bond with the enzyme substrate, hydrolyzing a peptide bond.
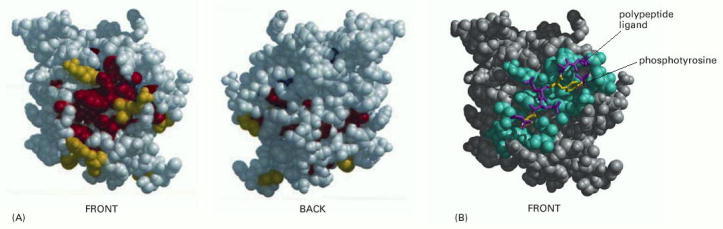
(A) Front and back views of a space-filling model of the SH2 domain, with evolutionarily conserved amino acids on the protein surface colored yellow, and those more toward the protein interior colored red. (B) The structure of the SH2 domain with its bound polypeptide. Here, those amino acids located within 0.4 nm of the bound ligand are colored blue. The two key amino acids of the ligand are yellow, and the others are purple. (Adapted from O. Lichtarge, H.R. Bourne, and F.E. Cohen, J. Mol. Biol. 257:342–358, 1996.)
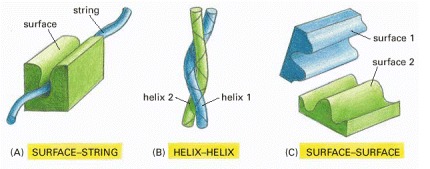
Only the interacting parts of the two proteins are shown. (A) A rigid surface on one protein can bind to an extended loop of polypeptide chain (a “string”) on a second protein. (B) Two α helices can bind together to form a coiled-coil. (C) Two complementary rigid surfaces often link two proteins together.
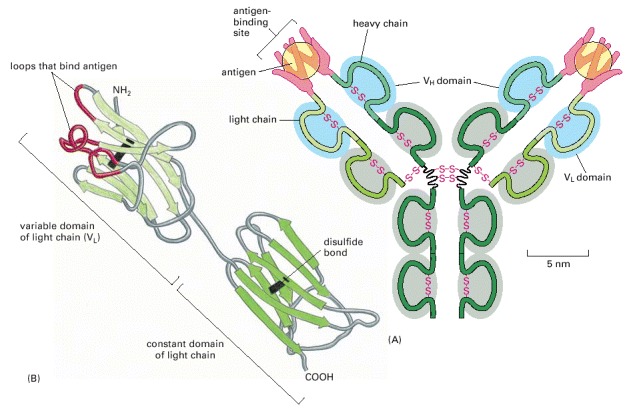
(A) A typical antibody molecule is Y-shaped and has two identical binding sites for its antigen, one on each arm of the Y. The protein is composed of four polypeptide chains (two identical heavy chains and two identical and smaller light chains) held together by disulfide bonds. Each chain is made up of several different immunoglobulin domains, here shaded either blue or gray. The antigen-binding site is formed where a heavy-chain variable domain (VH) and a light-chain variable domain (VL) come close together. These are the domains that differ most in their sequence and structure in different antibodies. (B) This ribbon model of a light chain shows the parts of the VL domain most closely involved in binding to the antigen in red. They contribute half of the fingerlike loops that fold around each of the antigen molecules in (A).
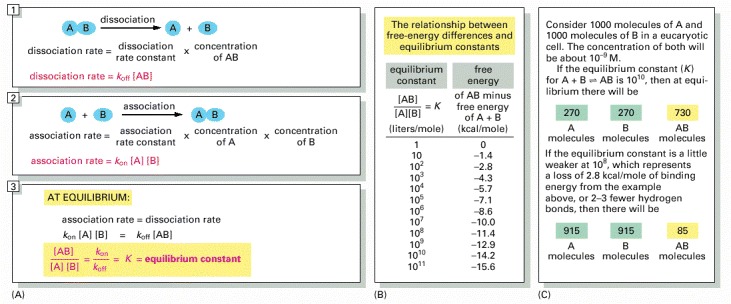
(A) The equilibrium between molecules A and B and the complex AB is maintained by a balance between the two opposing reactions shown in panels 1 and 2. Molecules A and B must collide if they are to react, and the association rate is therefore proportional to the product of their individual concentrations [A] × [B]. (Square brackets indicate concentration.) As shown in panel 3, the ratio of the rate constants for the association and the dissociation reactions is equal to the equilibrium constant (K) for the reaction. (B) The equilibrium constant in panel 3 is that for the association reaction A + B ↔ AB, and the larger its value, the stronger the binding between A and B. Note that, for every 1.4 kcal/mole of free-energy drop, the equilibrium constant increases by a factor of 10. (C) An example of the dramatic effect that the presence or absence of a few weak bonds can have in a biological context.
The equilibrium constant here has units of liters/mole: for simple binding interactions it is also called the affinity constant or association constant, denoted Ka. The reciprocal of Ka is called the dissociation constant, Kd (in units of moles/liter).
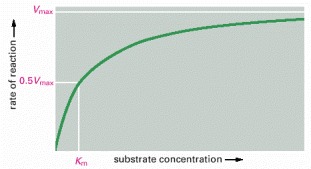
The rate of an enzyme reaction (V) increases as the substrate concentration increases until a maximum value (Vmax) is reached. At this point all substrate-binding sites on the enzyme molecules are fully occupied, and the rate of reaction is limited by the rate of the catalytic process on the enzyme surface. For most enzymes, the concentration of substrate at which the reaction rate is half-maximal (Km) is a measure of how tightly the substrate is bound, with a large value of Km corresponding to weak binding.
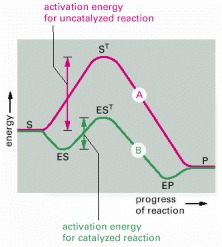
Often both the uncatalyzed reaction (A) and the enzyme-catalyzed reaction (B) can go through several transition states. It is the transition state with the highest energy (ST and EST) that determines the activation energy and limits the rate of the reaction. (S = substrate; P = product of the reaction.)
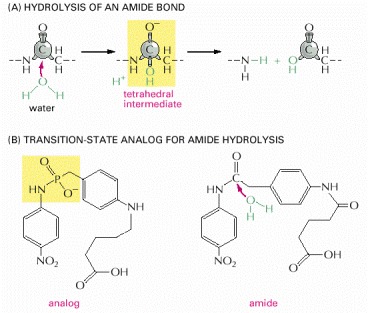
The stabilization of a transition state by an antibody creates an enzyme. (A) The reaction path for the hydrolysis of an amide bond goes through a tetrahedral intermediate, the high-energy transition state for the reaction. (B) The molecule on the left was covalently linked to a protein and used as an antigen to generate an antibody that binds tightly to the region of the molecule shown in yellow. Because this antibody also bound tightly to the transition state in (A), it was found to function as an enzyme that efficiently catalyzed the hydrolysis of the amide bond in the molecule on the right.
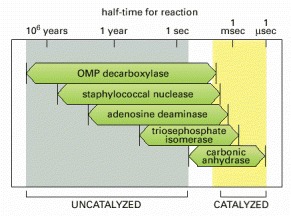
(Modified from A. Radzicka and R. Wolfenden, Science 267:90–93, 1995.)
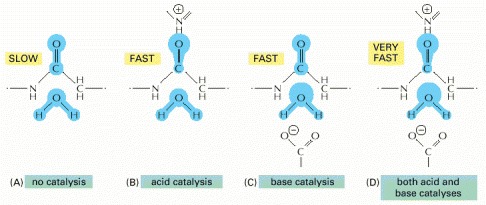
(A) The start of the uncatalyzed reaction shown in Figure 3-47A is diagrammed, with blue indicating electron distribution in the water and carbonyl bonds. (B) An acid likes to donate a proton (H+) to other atoms. By pairing with the carbonyl oxygen, an acid causes electrons to move away from the carbonyl carbon, making this atom much more attractive to the electronegative oxygen of an attacking water molecule. (C) A base likes to take up H+. By pairing with a hydrogen of the attacking water molecule, a base causes electrons to move toward the water oxygen, making it a better attacking group for the carbonyl carbon. (D) By having appropriately positioned atoms on its surface, an enzyme can perform both acid catalysis and base catalysis at the same time.
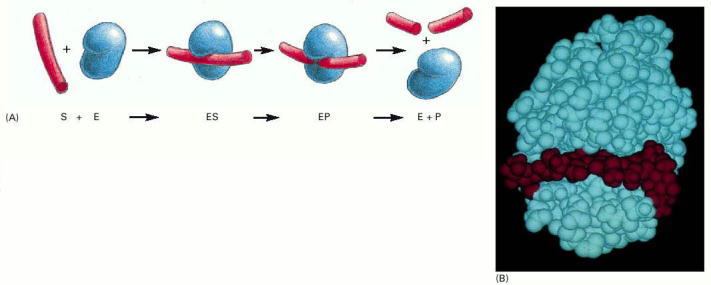
(A) The enzyme lysozyme (denoted as E) catalyzes the cutting of a polysaccharide chain, which is its substrate (S). The enzyme first binds to the chain to form an enzyme–substrate complex (ES) and then catalyzes the cleavage of a specific covalent bond in the backbone of the polysaccharide, forming an enzyme–product complex (EP) that rapidly dissociates. Release of the severed chain (the products P) leaves the enzyme free to act on another substrate molecule. (B) A space-filling model of the lysozyme molecule bound to a short length of polysaccharide chain before cleavage. (B, courtesy of Richard J. Feldmann.)
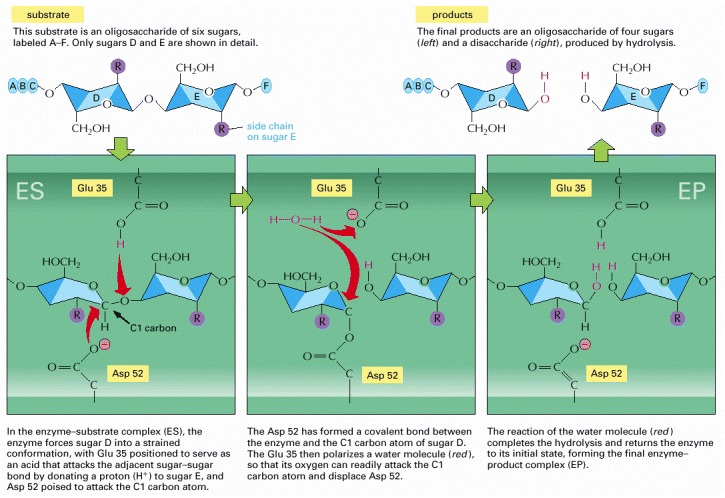
The top left and top right drawings depict the free substrate and the free products, respectively, whereas the other three drawings depict the sequential events at the enzyme active site. Note the change in the conformation of sugar D in the enzyme–substrate complex; this shape change stabilizes the oxocarbenium ion-like transition states required for formation and hydrolysis of the covalent intermediate shown in the middle panel. (Based on D.J. Vocadlo et al., Nature 412:835–838, 2001.)
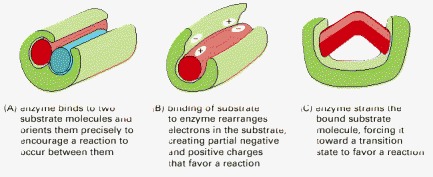
(A) Holding substrates together in a precise alignment. (B) Charge stabilization of reaction intermediates. (C) Altering bond angles in the substrate to increase the rate of a particular reaction.
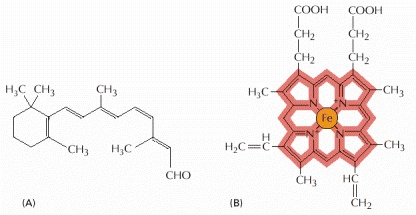
(A) The structure of retinal, the light-sensitive molecule attached to rhodopsin in the eye. (B) The structure of a heme group. The carbon-containing heme ring is red and the iron atom at its center is orange. A heme group is tightly bound to each of the four polypeptide chains in hemoglobin, the oxygen-carrying protein whose structure is shown in Figure 3-23.
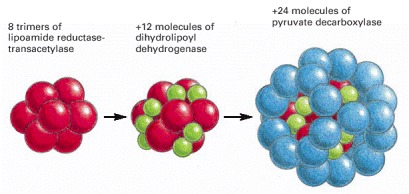
This enzyme complex catalyzes the conversion of pyruvate to acetyl CoA, as part of the pathway that oxidizes sugars to CO2 and H2O. It is an example of a large multienzyme complex in which reaction intermediates are passed directly from one enzyme to another.
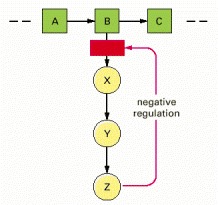
The end-product Z inhibits the first enzyme that is unique to its synthesis and thereby controls its own level in the cell. This is an example of negative regulation.
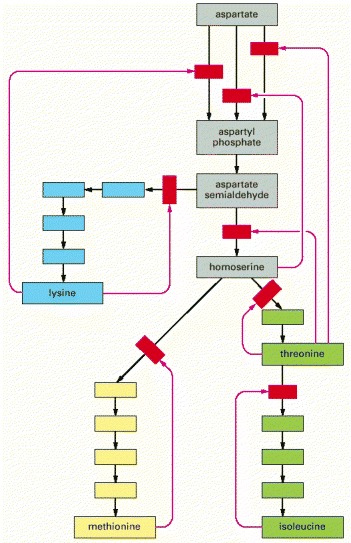
In this example, which shows the biosynthetic pathways for four different amino acids in bacteria, the red arrows indicate positions at which products feed back to inhibit enzymes. Each amino acid controls the first enzyme specific to its own synthesis, thereby controlling its own levels and avoiding a wasteful buildup of intermediates. The products can also separately inhibit the initial set of reactions common to all the syntheses; in this case, three different enzymes catalyze the initial reaction, each inhibited by a different product.
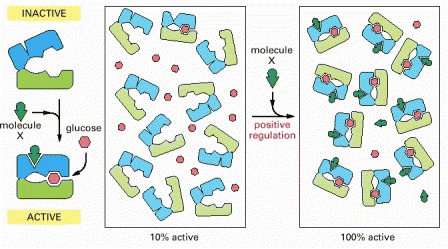
In this example, both glucose and molecule X bind best to the closed conformation of a protein with two domains. Because both glucose and molecule X drive the protein toward its closed conformation, each ligand helps the other to bind. Glucose and molecule X are therefore said to bind cooperatively to the protein.
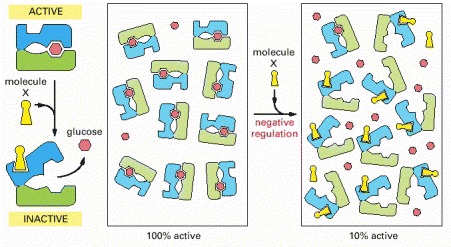
The scheme here resembles that in the previous figure, but here molecule X prefers the open conformation, while glucose prefers the closed conformation. Because glucose and molecule X drive the protein toward opposite conformations (closed and open, respectively), the presence of either ligand interferes with the binding of the other.
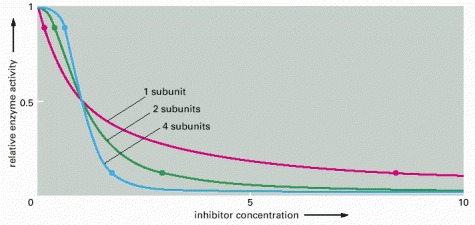
For an enzyme with a single subunit (red line), a drop from 90% enzyme activity to 10% activity (indicated by the two dots on the curve) requires a 100-fold increase in the concentration of inhibitor. The enzyme activity is calculated from the simple equilibrium relationship K = [I][P]/[IP], where P is active protein, I is inhibitor, and IP is the inactive protein bound to inhibitor. An identical curve applies to any simple binding interaction between two molecules, A and B. In contrast, a multisubunit allosteric enzyme can respond in a switchlike manner to a change in ligand concentration: the steep response is caused by a cooperative binding of the ligand molecules, as explained in Figure 3-60. Here, the green line represents the idealized result expected for the cooperative binding of two inhibitory ligand molecules to an allosteric enzyme with two subunits, and the blue line shows the idealized response of an enzyme with four subunits. As indicated by the two dots on each of these curves, the more complex enzymes drop from 90% to 10% activity over a much narrower range of inhibitor concentration than does the enzyme composed of a single subunit.
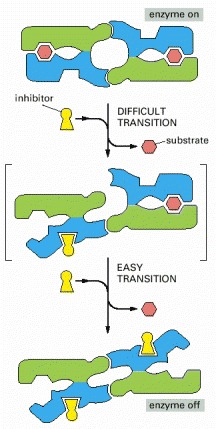
This diagram illustrates how the conformation of one subunit can influence that of its neighbor. The binding of a single molecule of an inhibitory ligand (yellow) to one subunit of the enzyme occurs with difficulty because it changes the conformation of this subunit and thereby destroys the symmetry of the enzyme. Once this conformational change has occurred, however, the energy gained by restoring the symmetric pairing interaction between the two subunits makes it especially easy for the second subunit to bind the inhibitory ligand and undergo the same conformational change. Because the binding of the first molecule of ligand increases the affinity with which the other subunit binds the same ligand, the response of the enzyme to changes in the concentration of the ligand is much steeper than the response of an enzyme with only one subunit (see Figure 3-59).
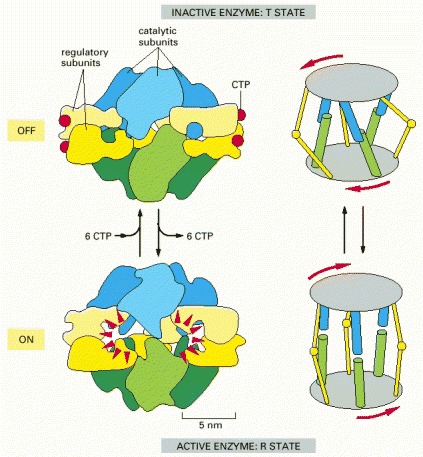
The enzyme consists of a complex of six catalytic subunits and six regulatory subunits, and the structures of its inactive (T state) and active (R state) forms have been determined by x-ray crystallography. The enzyme is turned off by feedback inhibition when CTP concentrations rise. Each regulatory subunit can bind one molecule of CTP, which is one of the final products in the pathway. By means of this negative feedback regulation, the pathway is prevented from producing more CTP than the cell needs. (Based on K.L. Krause, K.W. Volz, and W.N. Lipscomb, Proc. Natl. Acad. Sci. USA 82:1643–1647, 1985.)
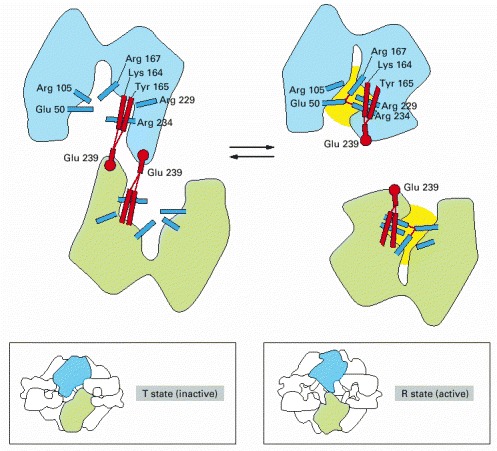
Changes in the indicated hydrogen-bonding interactions are partly responsible for switching this enzyme’s active site between active (yellow) and inactive conformations. Hydrogen bonds are indicated by thin red lines. The amino acids involved in the subunit–subunit interaction are shown in red, while those that form the active site of the enzyme are shown in blue. The large drawings show the catalytic site in the interior of the enzyme; the boxed sketches show the same subunits viewed from the enzyme’s external surface. (Adapted from E.R. Kantrowitz and W.N. Lipscomb, Trends Biochem. Sci. 15:53–59, 1990.)
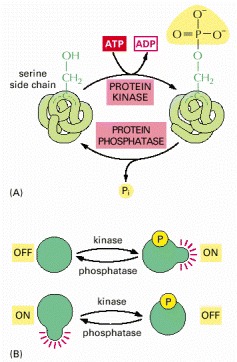
Many thousands of proteins in a typical eucaryotic cell are modified by the covalent addition of a phosphate group. (A) The general reaction, shown here, entails the transfer of a phosphate group from ATP to an amino acid side chain of the target protein by a protein kinase. Removal of the phosphate group is catalyzed by a second enzyme, a protein phosphatase. In this example, the phosphate is added to a serine side chain; in other cases, the phosphate is instead linked to the –OH group of a threonine or a tyrosine in the protein. (B) The phosphorylation of a protein by a protein kinase can either increase or decrease the protein’s activity, depending on the site of phosphorylation and the structure of the protein.
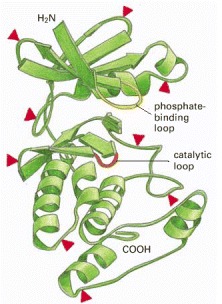
Superimposed on this structure are red arrowheads to indicate sites where insertions of 5–100 amino acids are found in some members of the protein kinase family. These insertions are located in loops on the surface of the enzyme where other ligands interact with the protein. Thus, they distinguish different kinases and confer on them distinctive interactions with other proteins. The ATP (which donates a phosphate group) and the peptide to be phosphorylated are held in the active site, which extends between the phosphate-binding loop (yellow) and the catalytic loop (orange). See also Figure 3-12. (Adapted from D.R. Knighton et al., Science 253:407–414, 1991.)
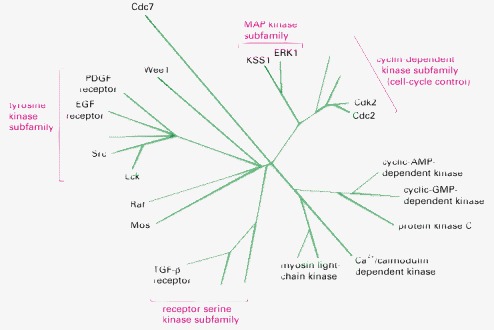
Although a higher eucaryotic cell contains hundreds of such enzymes, and the human genome codes for more than 500, only some of those discussed in this book are shown.
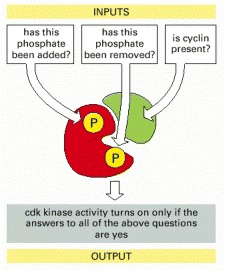
The function of these central regulators of the cell cycle is discussed in Chapter 17.
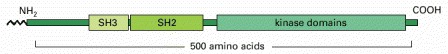
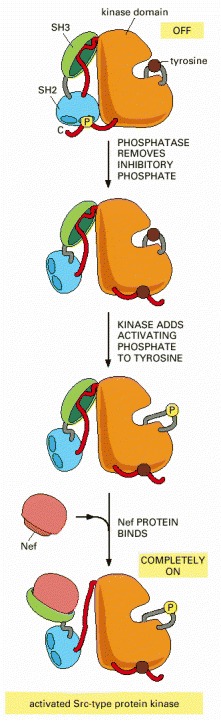
(Adapted from I. Moareti, et al., Nature 385:650–653, 1997.)
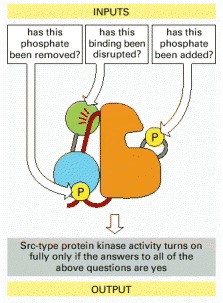
The disruption of the SH3 domain interaction (green) can involve replacing its binding to the indicated red linker region with a tighter interaction with the Nef protein, as illustrated in Figure 3-68.
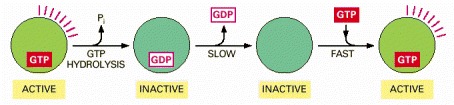
The activity of a GTP-binding protein (also called a GTPase) generally requires the presence of a tightly bound GTP molecule (switch “on”). Hydrolysis of this GTP molecule produces GDP and inorganic phosphate (Pi), and it causes the protein to convert to a different, usually inactive, conformation (switch “off”). As shown here, resetting the switch requires the tightly bound GDP to dissociate, a slow step that is greatly accelerated by specific signals; once the GDP has dissociated, a molecule of GTP is quickly rebound.
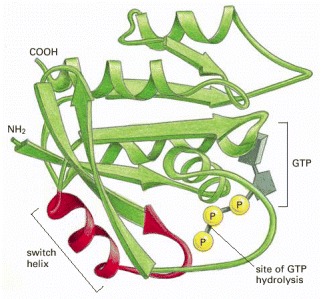
This monomeric GTPase illustrates the structure of a GTP-binding domain, which is present in a large family of GTP-binding proteins. The red regions change their conformation when the GTP molecule is hydrolyzed to GDP and inorganic phosphate by the protein; the GDP remains bound to the protein, while the inorganic phosphate is released. The special role of the “switch helix” in proteins related to Ras is explained next (see Figure 3-74).
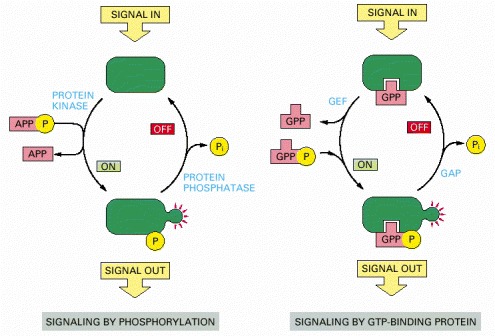
In both cases, a signaling protein is activated by the addition of a phosphate group and inactivated by the removal of this phosphate. To emphasize the similarities in the two pathways, ATP and GTP are drawn as APPP and GPPP, and ADP and GDP as APP and GPP, respectively. As shown in Figure 3-63, the addition of a phosphate to a protein can also be inhibitory.
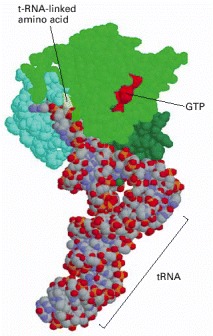
The three domains of the EF-Tu protein are colored differently, to match Figure 3-74. This is a bacterial protein; however, a very similar protein exists in eukaryotes, where it is called EF-1. (Coordinates determined by P. Nissen et al., Science 270:1464–1472, 1995.)
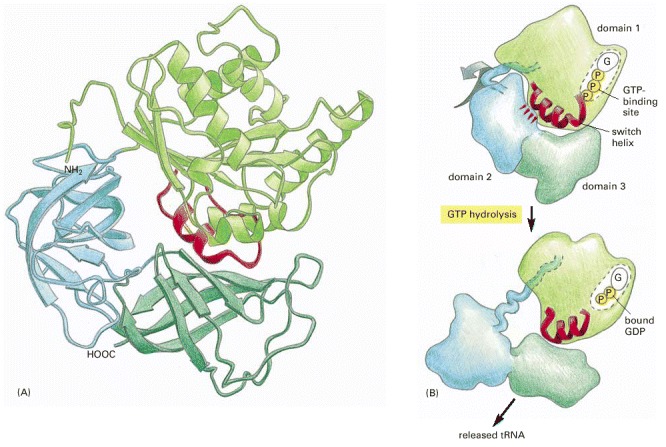
(A) The three-dimensional structure of EF-Tu with GTP bound. The domain at the top is homologous to the Ras protein, and its red α helix is the switch helix, which moves after GTP hydrolysis, as shown in Figure 3-71. (B) The change in the conformation of the switch helix in domain 1 causes domains 2 and 3 to rotate as a single unit by about 90° toward the viewer, which releases the tRNA that was shown bound to this structure in Figure 3-73. (A, adapted from H. Berchtold et al., Nature 365:126–132, 1993; B, courtesy of Mathias Sprinzl and Rolf Hilgenfeld.)
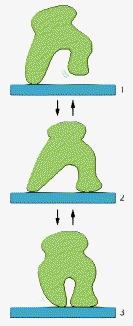
Although its three different conformations allow it to wander randomly back and forth while bound to a thread or a filament, the protein cannot move uniformly in a single direction.
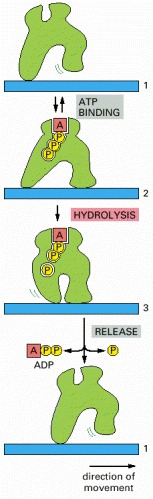
The transition between three different conformations includes a step driven by the hydrolysis of a bound ATP molecule, and this makes the entire cycle essentially irreversible. By repeated cycles, the protein therefore moves continuously to the right along the thread.
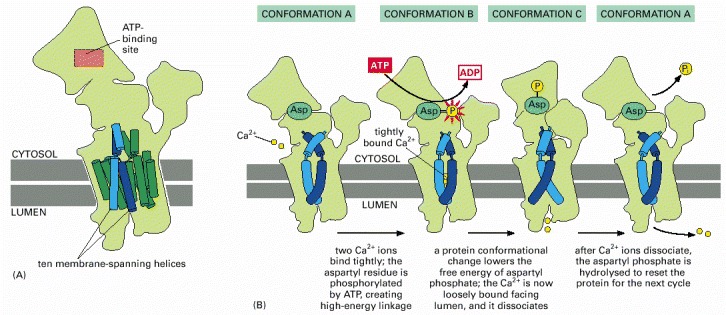
(A) The structure of the pump protein, formed from a single subunit of 994 amino acids (see Figure 11-15 for details). (B) A model for ion pumping. For simplicity, only two of the four transmembrane α helices that bind Ca2+ are shown.
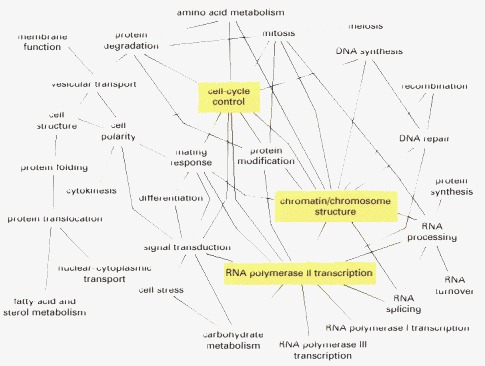
To produce this map, more than 1500 proteins were assigned to the indicated 31 functional groups. About 70 percent of the known protein–protein interactions were observed to occur between proteins assigned to the same functional group. However, as indicated, a surprisingly large number of interactions crossed between these groups. Each line in this diagram designates that more than 15 protein–protein interactions were observed between proteins in the two functional groups that are connected by that line. The three functional groups highlighted in yellow display at least 15 interactions with 10 or more of the 31 functional groups defined in the study. (From C.L. Tucker, J.F. Gera, and P. Uetz, Trends Cell Biol. 11:102–106, 2001.)