Chapter 23.1 - 23.2
1.) DNA Repair
Types and Consequences of DNA Damage
Unprogrammed chemical changes occur in all biological macromolecules because of
- environmental damage
- errors in synthesis.
For most biomolecules, including RNA, protein, and membrane lipids, the effects of these changes are minimized by turnover and replacement of the altered molecules.
DNA is distinctive, however, in that its information content must be transmitted virtually intact from one cell to another during cell division or the reproduction of an organism.
- Thus, DNA requires metabolic stability.
This stability is maintained in two ways
- By a highly accurate replication process
- By mechanisms for correcting genetic mistakes when DNA suffers damage.
In Chapter 22 we described mechanisms used to ensure high replication fidelity.
Here we discuss the kinds of environmental and endogenous damage that occur and the processes that repair the damage.
Figure 23.2 identifies the major kinds of DNA damage that arise through either replication errors or the actions of environmental agents.
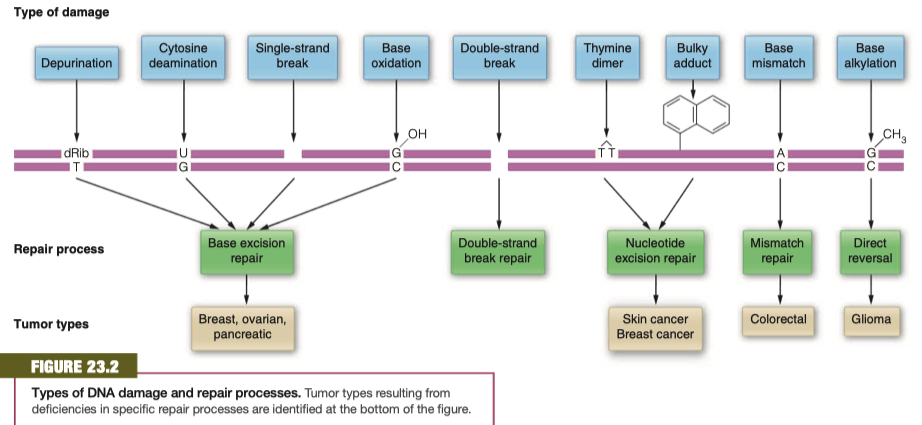
Connection One of the principal causes of cancer is genomic instability resulting from the defective repair of DNA damage.
The figure also identifies each of the major processes that can repair particular types of damage.
Because unrepaired damage is mutagenic and cancer arises from genomic instability (Chapter 20), the figure also identifies forms of cancer that result from defects in particular repair processes.
Some of the most prominent endogenous DNA-damaging reactions include :
- (1) depurination, resulting from cleavage of the glycosidic bond between deoxyribose and a purine base;
- (2) deamination, usually the hydrolytic conversion of a DNA-cytosine residue to uracil
- (3) oxidation, notably the oxidation of guanine to 8-oxoguanine or of thymine to thymine glycol
- (4) nonenzymatic methylation by S-adenosylmethionine.
- Figure 23.3 shows the frequency with which they occur in mammalian cells.
Environmental DNA-damaging agents include :
- ionizing radiation
- ultraviolet radiation
- DNA methylating reagents, such as N-methylN′-nitro-N-nitrosoguanidine (MNNG; next page)
- DNA crosslinking reagents, such as the anticancer drug cisplatin
- Bulky hydrocarbons, such as benzo[a]pyrene, one of the carcinogenic agents in tobacco smoke.
As we discuss the repair of spontaneous or environmental DNA damage, we will see that some repair processes are accurate (i.e., the original DNA sequence is restored), while some lead to mutations (i.e., the repair process is inaccurate).
As mentioned above and in Chapter 20, cancer results from an accumulation of somatic cell mutations.
- Therefore, the mechanisms of DNA repair are under intense study as determinants of an animals susceptibility to cancer.
Our earliest understanding of DNA repair came from study of the lethal and mutagenic effects of ultraviolet light on bacteria or viruses.
How do we know that DNA is the target for UV light?
- Organisms were found to be killed or mutagenized most efficiently by light of wavelength 260 nm, the absorption maximum for DNA.
- Analysis of the photoproducts, or altered DNA constituents after irradiation, showed the most prominent to be intrastrand dimers consisting of two bases (usually thymine) joined either by a cyclobutane ring involving carbons 5 and 6 or by a bond from C4 of one pyrimidine to C6 of the next (Figure 23.4).
- These thymine dimers were identified early as biologically significant photoproducts because the relative abundance of thymine dimers in irradiated DNA correlated most closely with death or mutagenesis of irradiated phages or bacteria.
- Thus, the ability of an organism to survive ultraviolet irradiation was partially due to its ability to remove thymine dimers from its DNA.
- The cyclobutane structure draws the adjacent thymine residues together, distorting the helix in a way that blocks replication past this site, while the 6–4 dimer is strongly mutagenic because it induces inaccurate replication.
Concept Cyclobutane thymine dimer is the most lethal photoproduct in UV light–irradiated DNA, and 6–4 dimer is the most mutagenic
2.) Direct repair of Damaged DNA Bases: Photoreactivation and Alkyltransferases
- Of the half dozen well-understood DNA repair processes, most involve removal of the damaged nucleotides, along with several adjacent residues, followed by replacement of the excised region using information encoded in the complementary (undamaged) strand.
- However, at least two processes involve reactions that directly change the damaged bases, rather than removing them.
Concept DNA can be repaired directly, by changing a damaged base to a normal one, or indirectly, by replacing a DNA segment containing the damaged nucleotide.
Photoreactivation
- In some organisms, not including mammals, DNA damage can be repaired directly, by visible light irradiation at about 370 nm, in a process called photoreactivation.
- This involves the action of the enzyme DNA photolyase.
- The enzyme binds to DNA in a light-independent process, specifically at the site of pyrimidine dimers.
- In the presence of visible-wavelength light, the bonds linking the pyrimidine rings are broken, after which the enzyme can dissociate in the dark.
- Clues to the photolyase mechanism have come with the finding that the enzyme contains two chromophores.
- One chromophore is bound flavin adenine dinucleotide, deprotonated and in the reduced state (FADH−)
- The second in many photolyases is 5,10-methenyltetrahydrofolate (introduced in Chapter 18).
- Mechanistic studies suggest a process akin to photosynthesis, with the second chromophore functioning as a light-harvesting factor and transmitting light energy by fluorescent resonance energy transfer to FADH−.
- Hence, FADH− functions like the photochemical reaction center, transferring an electron to the dimer and breaking the pyrimidine–pyrimidine bonds by a free radical mechanism.
- The crystal structure of the E. coli photolyase shows 5,10-methenyltetrahydrofolate bound at the surface, between the N-terminal and C-terminal domains, with FADH− bound deeply within the C-terminal domain (Figure 23.5a).
- Figure 23.5b shows the probable reaction pathway.
O6 - Alkylguanine Alkyltransferase
- Like UV irradiation, exposure of DNA to a methylating or ethylating reagent yields modified DNA bases, some of which cause mutagenesis or death if not repaired.
- Some alkylating agents are used in cancer chemotherapy because of their ability to block DNA replication and, hence, cell proliferation.
- Some alkylating agents are used as mutagens in the laboratory, including N-methyl-N′-nitro-N-nitrosoguanidine (MNNG, page 717) and others shown here.
- MNNG and other alkylating agents react primarily with purines.
- The most highly mutagenic of these products, O6-methylguanine, has a high probability of pairing with thymine when the modified strand replicates (Figure 23.6).
- Thus, alkylation of a DNA-guanine stimulates a GC → AT transition mutation (see Figure 23.6b) where mG is a methylguanine residue.
- Repair of this type of damage involves action of O6-alkylguanine alkyltransferase, which transfers a methyl or ethyl group from an O6-methylguanine or O6-ethylguanine residue to a cysteine residue in the active site of the protein.
- Remarkably, this “enzyme,” which is widely distributed in bacteria and eukaryotes, can function only once.
- Having become alkylated, it cannot remove the alkyl group, and the protein molecule cannot repeat the process.
- However, the alkylated form of the protein is a transcriptional activator, which stimulates transcription of the gene encoding the alkyltransferase.
- It thereby allows the cell to adapt to alkylation damage by using the alkylated protein to signal the cell to produce more of the protein needed to repair the damage.
Concept Direct repair enzymes include photolyase, which uses light energy to pyrimidine dimers, and alkyltransferases, “enzymes” that are inactivated after just one catalytic cycle.
3.) Nucleotide Excision Repair: Excinucleases
- is an enzyme system capable of repairing thymine dimers created in DNA by UV irradiation.
- Unlike photoreactivation, this process can take place in the dark.
- The enzyme system involved, which in E. coli includes the products of genes uvrA, uvrB, and uvrC, also acts upon a wide range of other DNA lesions that may be quite bulky, such as those created by large alkyl or aryl groups that distort the DNA double helix.
- Similar systems exist in mammalian cells and in yeast, so the process is probably universal.
- As shown in Figure 23.7 for E. coli, the three-subunit UvrABC enzyme recognizes a lesion (a thymine dimer in the example shown) and, with the help of ATP hydrolysis, forces DNA to bend, step 1.
- Dissociation of UvrA and its replacement by UvrC, step 2, leads to cleavage of the damaged strand at two sites—seven nucleotides to the 5′ side of the damaged site and four nucleotides to the 3′ side.
- The resulting gap in the damaged DNA strand is ~11 nucleotides in length, with a 3′ hydroxyl group and a 5′ phosphate at the ends, step 3.
- Polymerase and ligase action then replaces the damaged 11-mer with new DNA, using the undamaged DNA strand as a template, step 4.
- Helicase II, the product of the uvrD gene, is also required, presumably to unwind and remove the excised oligonucleotide, which is ultimately broken down by other enzymes.
- Because 11 nucleotides amount to about one turn of the DNA helix, the enzyme may catalyze both cleavages from the same DNA binding site.
- The UvrABC enzyme is not a classical endonuclease because it cuts at two distinct sites
- The term excinuclease has been proposed, denoting its role in excision repair.
- This system is also involved in repairing the DNA damage that results when two strands covalently crosslink to each other.
Concept Excision repair involves endonuclease cleavage on both sides of a damaged site, followed by replacement synthesis.
- Excision repair also occurs in mammalian cells, as shown by the presence of a human excinuclease that cleaves at positions −22 and +6 relative to a thymine dimer.
- Unlike bacteria, the process in humans requires two different endonucleases—one cutting on the 5′ side and one on the 3′ side.
- Excision repair in humans originally came to light through studies of a rare genetic disease called xeroderma pigmentosum (XP).
- XP is actually a family of diseases in which one or more enzymes of the excision pathway are deficient.
- In affected humans there is no known way to treat the condition.
- The biological consequences of XP include extreme sensitivity to sunlight and a high incidence of skin cancers.
- Although overexposure to the ultraviolet rays in sunlight increases the risk of skin cancer for all humans, the greatly increased frequency of skin cancer in XP patients highlights the importance of UV repair pathways to mammals.
Connection Serious clinical consequences, notably, enhanced cancer susceptibility, result from genetic abnormalities of NER.
- Later studies of nucleotide excision repair showed that active genes (those undergoing transcription) are preferred substrates for excision repair, and within these genes the template DNA strand is preferentially repaired.
- This transcription-coupled repair may initiate when a transcribing RNA polymerase becomes stalled at the site of a DNA lesion.
- Coupling transcription with repair helps to ensure the integrity of genes that are actually being used.
- In mammalian cells, transcription-coupled repair is a specialized mode of NER that requires additional proteins.
- Cockayne’s syndrome results from genetic defects in one or more of the human enzymes of transcription-coupled repair.
- Children with this condition age prematurely and usually die of aging-related symptoms by age 12.
- Another human gene, BRCA1, discovered by Mary-Claire King, is also implicated in transcription-coupled NER.
- Mutations in BRCA1, and the related BRCA2 gene, greatly increase the risk of breast and ovarian cancer.
- The BRCA genes became more widely known to the public in 2013, when actress Angelina Jolie underwent a preventive double mastectomy because she carried BRCA mutations and had a family history of breast cancer deaths.
- As noted earlier, NER also corrects DNA that has been damaged by the formation of bulky DNA adducts.
- Many environmental carcinogens give rise to such adducts.
- In the absence of repair, or following error-prone repair, these adducts can lead to mutations.
- For example, polycyclic aromatic hydrocarbons (PAHs) are prevalent organic pollutants formed by incomplete combustion (e.g., by burning charcoal in a backyard grill or by smoking).
- As with many environmental carcinogens, PAHs are not directly reactive with DNA.
- Indeed, their lack of chemical reactivity accounts for their persistence in the environment.
- However, PAHs can undergo metabolic activation (i.e., biotransformation), thus leading to reactive electrophilic intermediates that can bind to DNA and other macromolecules.
- These reactions can occur in the liver and in other organs.
- The best-characterized route for the metabolic activation of PAHs is the pathway for benzo[a]pyrene metabolism (Figure 23.8).
- First, oxygenation catalyzed by a cytochrome P450 generates an epoxide intermediate.
- This epoxide is hydrolyzed, in a reaction catalyzed by epoxide hydrolase, to give a dihydrodiol product.
- Finally, a second cytochrome P450-catalyzed oxygenation produces the highly reactive benzo[a]pyrene 7,8-dihydrodiol-9,10-epoxide (BPDE).
- BPDE is highly reactive with DNA, forming bulky covalent adducts primarily, but not exclusively, with guanine.
Connection The carcinogenicity of polycyclic aromatic hydrocarbons (PAHs) stems from their ability to react with DNA bases and form mutagenic intermediates.
4.) Base excision repair: DNA N-Glycosylases
- In addition to NER, there is another form of excision repair, base excision repair (BER).
- Like NER, BER also removes one or more nucleotides from a site of base damage.
- However, this process begins with enzymatic cleavage of the glycosidic bond between the damaged base and deoxyribose.
Replacement of Uracil in DNA by BER
- One of the best-understood BER systems scans DNA to remove uracil.
- Uracil can base-pair with adenine in a DNA duplex, and DNA polymerases can readily accept deoxyuridine triphosphate as a substrate in place of thymidine triphosphate.
- Yet cells possess an elaborate two-stage process that prevents dUMP residues from accumulating in DNA.
- The first stage, described in Chapter 19, involves cleavage by dUTPase of dUTP to dUMP, thereby minimizing the dUTP pool and its use as a replication substrate.
- The second stage involves uracil-DNA N-glycosylase (Ung), an enzyme that removes any dUMP residues in DNA that might have arisen either through deamination of a dCMP residue or through incorporation of a dUTP that escaped the action of dUTPase.
Concept A base excision repair process removes uracil residues in DNA, whether they arose through the deamination of cytosine residues or the incorporation of deoxyuridine nucleotides instead of thymidine nucleotides.
- As shown in Figure 23.9, Ung hydrolytically cleaves the glycosidic bond between N-1 of uracil and C-1′ of deoxyribose.
- This yields free uracil and DNA with an apyrimidinic site (i.e., a sugar residue lacking an attached pyrimidine).
- Another enzyme, apyrimidinic endonuclease (AP endonuclease), recognizes this site and cleaves the phosphodiester bond on the 5′ side of the deoxyribose moiety.
- This is followed in bacteria by the nick translation activity of DNA polymerase I (Figure 22.14), which inserts dTTP as a replacement for the dUMP that was removed, displacing the deoxyribose phosphate residue in the AP site.
- This is removed by a deoxyribose-5′-phosphatase.
- The resulting nick is sealed by DNA ligase.
- Ung, like all DNA glycosylases examined to date, acts by flipping the target base out of the DNA duplex and binding it in a pocket where cleavage occurs.
- Structural analysis of human UNG shows that the pocket is small enough to exclude purine bases.
- More important, as shown in Figure 23.10, the pocket excludes thymine because of negative steric interaction between the thymine methyl group at C5 and Tyr-147. (By convention, eukaryotic genes and gene products are denoted with capital letters; thus, UNG is the human gene that encodes the protein UNG. In bacteria, gene ung encodes protein Ung.)
- Why do cells go to all this trouble just to replace a nucleotide that does not affect the information encoded in DNA?
- Almost certainly, U-A base pairs are not the true target of this DNA repair system.
- Uracil residues in DNA also arise through spontaneous deamination of cytosine residues (Figure 23.3).
- This alteration does change the genetic sense because it converts a G-C base pair to a G-U pair, and in a subsequent round of replication the U-containing strand would give rise to an A-T base pair—a GC-to-AT transition (change of a purine-pyrimidine pair to another purine-pyrimidine).
- The uracil repair system prevents this mutation but does not discriminate between uracils paired with adenines or with guanines.
- Consistent with this model is the hypermutable phenotype displayed by mutant bacteria lacking an active Ung.
- Such strains exhibit elevated rates of spontaneous mutagenesis, resulting from the accumulation of DNA dUMP residues paired with dGMP.
- The replacement of U by T in DNA is one example of BER.
- Another example, described next, involves repair of damage caused by reactive oxygen species.
Repair of Oxidative Damage to DNA
- Most cells contain several DNA-N-glycosylases, including those specific for the alkylated bases N-methyladenine, 3-methyladenine, and 7-methylguanine.
- Of particular interest is the BER process used to repair oxidative DNA damage.
- As was indicated in Figure 23.2, one of the most abundant of the numerous oxidation products resulting from DNA exposure to ROS is 8-oxoguanine.
- This is a strongly mutagenic alteration because 8-oxoguanine pairs readily with adenine.
- Oxidation of a G paired with C gives rise to a C-oxoG base pair.
- A subsequent round of replication readily gives rise to an A-oxoG pair, which then becomes A-T in the next round.
- Hence, the C-oxoG mispair can be an intermediate in a transversion mutation C‐G → C‐oxoG → A‐oxoG → A‐T. (A transversion changes a pyrimidine-purine base pair to a purine-pyrimidine pair.)
- In E. coli, three genes encode proteins that minimize oxidative mutagenesis: mutM, mutY, and mutT. (The “mut” designation indicates that mutations in any one of these genes have the effect of increasing the spontaneous mutation rate.)
- All three gene products have mammalian homologs. MutM and MutY are both DNA-base glycosylases.
- MutM (or its mammalian homolog, OGG1), which we describe here, cleaves 8-oxoguanine from DNA to initiate BER.
- The repair process, similar to DNA-uracil repair, results in replacement of oxoG by G.
- Like UNG, described earlier, human OGG1 DNA glycosylase distinguishes target oxoG bases from nontarget bases such as guanine by excluding nontarget bases from the active site where cleavage occurs.
- Structural analysis of human OGG1 revealed that 8-oxoguanine is able to enter the binding pocket, while guanine is completely excluded due to both attractive and repulsive forces (Figure 23.11).
5.) Mismatch Repair
- Mismatches, or non-Watson–Crick base pairs in a DNA duplex, can arise through replication errors, through deamination of 5-methylcytosine in DNA to yield thymine, or through recombination between DNA segments that are not completely homologous.
- In addition, mismatches result when DNA polymerase encounters a short repeated sequence and slides along its template, creating short loops or bulges in duplex DNA (see Figure 22.7).
- We best understand the correction of replication errors, so that is what we describe here.
- If DNA polymerase introduces an incorrect nucleotide, creating a non-Watson–Crick base pair, the error is usually corrected by the polymerase-associated 3′ exonuclease activity (Chapter 22).
- If the error is not corrected immediately, the fully replicated DNA will contain a mismatch at that site.
- This error can be corrected by mismatch repair.
- In E. coli, the proteins that participate include the products of genes mutH, mutL, and mutS, plus an enzyme called helicase II.
- The mismatch correction system scans newly replicated DNA, looking for both mismatched bases and single-base insertions or deletions.
- MutS binds to DNA at the site of the mismatch, followed by the binding of MutL and then MutH. MutS is a “motor protein,” which uses the energy of ATP hydrolysis to pull DNA from both directions until it reaches the site at which repair is to begin.
- When it finds an appropriate signal, part of one strand containing the mismatched region is cut out and replaced (Figure 23.12).
Concept The mismatch repair system in bacteria uses DNA methylation to identify the strand that has a mispaired nucleotide.
- How does the mismatch repair system recognize the correct strand to repair?
- If it chose either strand randomly, it would choose incorrectly half the time and there would be negligible gain in replication accuracy.
- The mismatch repair enzymes identify the newly replicated strand because for a short period that DNA is unmethylated.
- In E. coli, the sequence –GATC– is crucial because that is the site methylated soon after replication, by the product of the dam gene (DNA adenine methylase).
- The mismatch repair enzymes look for –GATC– sequences that are not methylated.
- Recognition of an unmethylated GATC can target that strand for mismatch correction at a site as far as 1 kbp between the mismatch and the GATC site, in either direction.
- Once the methylation system has acted on all GATC sites in the daughter strand, it is too late for the mismatch repair system to recognize the more recently synthesized DNA strand, and any advantage in total DNA replication fidelity is lost.
- When the system functions properly, it has the effect of increasing overall replication fidelity by about 100-fold—from about 1 error in 107 base pairs replicated to about 1 in 109 or better.
- As shown in Figure 23.12, the MutHLS complex moves along DNA in both directions until it encounters the nearest 5′-GATC sequence.
- An endonuclease activity of MutH then cleaves on the 5′ side of the G in the unmethylated strand.
- At that point helicase II unwinds the DNA, moving back past the mismatch, followed by an exonuclease that digests the displaced single strand.
- The resultant gap is filled by DNA polymerase III holoenzyme and DNA ligase, working in concert with SSB.
A similar mismatch repair system exists in eukaryotic cells.
The mismatch recognition step is somewhat more complex, however, because three different MutS homologs (MSH proteins) are involved—MSH2, MSH3, and MSH6.
These three proteins form heterodimers, with specificities for different types of mismatches.
Still unknown is the mechanism by which eukaryotic systems recognize and initiate repair on the newly replicated DNA strand;
- it is clear that DNA methylation is not involved.
Recently, it has been proposed that ribonucleotides incorporated by imperfect specificity of DNA polymerase (Chapter 22) could provide such a signal.
In the parental strands, any ribonucleotide incorporation would have been repaired.
- Just as is seen with bacteria, mutations in eukaryotic genes that control mismatch repair confer a mutator phenotype, raising spontaneous mutation rates at all loci.
- How do such mutations affect the biology of human cells?
- As mentioned in Chapter 20, the progression of a normal cell to a cancer cell involves the accumulation of multiple mutations in tumor cell precursors.
- Cells lacking mismatch repair activity do have elevated mutation rates, so it was a logical development when mutations in mismatch repair genes were found in tumor cells from individuals with a heritable cancer predisposition called HNPCC (heritable nonpolyposis colon cancer).
- To date, germ-line mutations in the genes for five different mismatch repair proteins have been found to be associated with HNPCC.
- The most common forms of the disease involve mutations affecting either hMLH1 (human Mut L homolog) or hMHS2 (human MutS homolog).
- The cancer predisposition is inherited in an autosomal dominant fashion, suggesting that most affected individuals are heterozygous, with one wild-type and one nonfunctional allele.
- Mismatch repair is normal until a somatic cell mutation inactivates the one functional allele and mismatch repair capacity is essentially abolished, with a consequent increase in spontaneous mutagenesis.
- It is unclear why mutations affecting mismatch repair are associated specifically with tumors in the colon.
Connection Genetic deficiencies in mammalian mismatch repair are often associated with colorectal cancer.
- Tumor cells from those affected with HNPCC exhibit microsatellite instability—a phenomenon in which there are large numbers of mutations in regions of the genome containing repeats of single-, double-, and triple-nucleotide sequences, usually with large increases in the numbers of repeating units in such sequences.
- These data suggest that the product and template strands can normally slip at such sites, so that DNA polymerase copies a short repeating sequence more than once, or else skips a segment.
- This creates a heteroduplex with a short loop, as we showed in Chapter 22 (Figure 22.7) for replication errors leading to Huntington’s disease.
- Normally, a replication error of this type would be corrected by mismatch repair, but such errors persist and accumulate in a cell lacking normal mismatch repair.
- Studies of this type have given scientists insight into the nature of cancer as a progressive genetic disease.
6.) Double-Strand Break Repair
- A double-strand break (DSB), a lesion that can be caused by ionizing radiation or by replication stalling, is the most lethal form of DNA damage because it destroys the physical integrity of a chromosome.
- DSBs occur about 50 times per mammalian cell cycle.
- Moreover, double-stranded DNA breaks occur naturally during meiotic recombination.
- DSBs can be repaired by either of two processes—homologous recombination (HR), using sequence information from an undamaged sister chromatid, or nonhomologous end joining (NHEJ). (From the standpoint of evolution, homologous means “derived from a common ancestor,” but we use it in a broader sense, meaning “having similar sequences.”)
- NHEJ is more efficient, but if DNA ends are not rejoined at the precise sites where breakage occurred, genetic information becomes lost or scrambled.
- By contrast, HR normally repairs the broken site precisely, but it can occur only during the S or G2 cell-cycle phases, when a homologous chromosome is available.
- Both processes begin with several signaling proteins associating at the severed ends.
- A principal player in vertebrates is a protein kinase called ATM (ataxia telangiectasia mutated).
- An early event in both HR and NHEJ is phosphorylation of a variant form of H2 histone, H2AX, in nucleosomes near the break.
- This may be part of a chromatin remodeling process that helps to move core particles out of the way, exposing DNA ends for processing.
- Double-strand break repair is better understood for homologous recombination, and that is what we outline here.
- Although it is simple to visualize conceptually, as shown in Figure 23.13,
- HR is a complex process, involving extensive signaling reactions as part of the DNA damage response (see page 725).
- As studied in yeast, a signaling complex called ATM activates a nuclease in another complex called MRN, as well as signaling downstream effectors via an associated protein kinase activity.
- The MRN-associated nuclease trims away 5′ DNA ends, leaving 3′-terminated single-stranded ends, which become coated with RPA (replication protein A; single-strand binding protein).
- The BRCA2 protein (page 720) is involved in this process.
- RNA polymerase II transcribes the single-stranded DNA ends, and ribonuclease H degrades the RNA in the hybrid.
- RPA then coats the 3′ DNA ends.
- Not shown is the binding of Rad51, a protein that scans the single-stranded DNA ends and the homologous undamaged chromosome, seeking regions of sequence homology, thereby facilitating base pairing and subsequent ligation.
- Rad51 is the eukaryotic counterpart of bacterial RecA, whose action is described on page 728.
Connection Genetic defects involving double-stranded break repair, particularly in genes BRCA1 and BRCA2, are risk factors for breast or ovarian cancer.
- Human mutations are known that affect several of the proteins involved in DSB repair.
- As mentioned previously, mutations in either BRCA1 or BRCA2 are risk factors for developing breast or ovarian cancer.
- BRCA2 mutations are also associated with Fanconi’s anemia, a rare condition characterized by bone marrow failure.
- ATM received its name from the deficiency syndrome ataxia telangiectasia, which involves premature aging, cerebellar degeneration, and enhanced cancer susceptibility.
Concept Double-strand DNA breaks can be repaired either by homologous recombination (HR) or by nonhomologous end joining (NHEJ), a process that does not require DNA sequence homology at the ends being joined.
7.) Daughter-Strand Gap Repair
- Bacteria also use HR as a way to repair DNA damage.
- However, because bacteria have but a single chromosome, the undamaged duplex needed as a repair template is the replicated portion of an incompletely replicated chromosome.
- This process, called daughter-strand gap repair, comes into play if DNA damage is sufficient to exceed the cell’s capacity for photoreactivation or excision repair.
- The process is schematized in Figure 23.14.
- When a replisome encounters a thymine dimer or a bulky lesion, it cannot replicate past this site.
- A recombination-related process can take place, not to repair the damage, but to allow replication to continue, saving the damaged site for subsequent repair by another process.
- Daughter-strand gap repair depends on the RecA protein mentioned earlier.
- RecA, like its eukaryotic counterpart, Rad51, catalyzes strand pairing, or strand assimilation—the joining of two different DNAs by homologous base pairing.
8.) Translesion Synthesis and the DNA Damage Response
- With the exception of direct-reversal repair processes like photoreactivation, DNA repair requires the action of a polymerase to fill in a single-strand gap in the DNA duplex.
- If the gap contains a damaged structure, like a thymine dimer or a bulky lesion, a replicative polymerase is unable to copy past the damaged site, and the repair process aborts.
- A new set of damage-tolerant DNA polymerases, inducible by DNA damage, comes into play.
- Recall from Chapter 22 that bacteria contain five DNA polymerases, only two of which are involved in replication, while eukaryotic cells have as many as nine damage-inducible polymerases.
- Most of these enzymes have low fidelity, partly because they lack 3′ exonucleolytic proofreading and partly because their catalytic sites are larger and more open than those of high-fidelity replicative polymerases.
- The enzymes are inducible as a result of DNA damage, but the control mechanisms differ markedly between bacterial and eukaryotic organisms.
- The SOS response in bacteria is a metabolic alarm system that helps the cell to save itself in the presence of potentially lethal stresses.
- Inducers of the SOS response include DNA damaging agents such as ultraviolet irradiation, thymine starvation, and inactivation of genes essential to DNA replication.
- Responses include mutagenesis, filamentation (in which cells elongate by growth but don’t divide), and activated excision repair. Mutagenesis occurs because, under SOS conditions, the gaps that are formed are filled by inaccurate DNA polymerases.
- In fact, this process is the principal pathway by which ultraviolet light stimulates mutagenesis in bacteria.
- Induction of the SOS response by UV light or other DNA damage activates transcription of about 40 genes in E. coli.
- We discuss the mechanism of transcriptional activation in Chapter 26.
- Among these SOS-inducible genes are those encoding DNA polymerases IV and V.
- Both of these enzymes are highly error-prone.
- After SOS activation, the processing of two gene products (UmuC and UmuD) gives rise to a pol V molecule.
- This associates with RecA bound to ATP, giving the active form of the enzyme.
- When the replisome containing pol III holoenzyme stalls at a damage site, pol V replaces it and binds to the sliding clamp.
- Once the damaged site has been copied (inaccurately), pol V somehow steps aside, and pol III holoenzyme completes the replication process. Pol IV functions similarly, but we know less about it.
Concept DNA can be repaired after replication, either by recombination or by inducible error-prone repair. Both processes require RecA.
- Considering that most mutations are probably deleterious, what is the advantage to the cell of accumulating mutations during DNA repair?
- Probably none, except that the alternative would be for the cell to die.
- In other words, extensive mutagenesis is a worthwhile price for the cell to pay, if a massive UV dose has overwhelmed all other repair pathways and if error-prone replication past a dimer is necessary for the cell to survive.
- As noted earlier, translesion synthesis is inducible in eukaryotic cells as well.
- Recall from Chapter 22 that eukaryotic cells were formerly thought to have five DNA polymerases.
- Since the late 1990s, a remarkable variety of new DNA polymerases has been discovered in eukaryotic cells, and the total number of human DNA polymerases now stands at 15.
- Four of the newly discovered polymerases—Pol η, Pol ι, Pol κ, and REV1—are specialized to replicate past specific types of DNA damage.
- These enzymes all have generally low fidelity, with the exception of pol η, which can replicate past thymine dimers in an essentially error-free manner.
- Induction of some or all of the repair polymerases in eukaryotic cells is part of the DNA damage response, a complex set of events involving a number of signaling pathways, which we introduced on page 725.
- At least 700 proteins undergo phosphorylation during this process, catalyzed by ATM and ATR and other kinases.
- Depending on the severity of DNA damage, the cell type, and the phase of the cell cycle, the cellular responses may activate DNA repair pathways, cell-cycle arrest, or programmed cell death (apoptosis).
- We have previously mentioned p53 (Chapter 20), an effector that acts as a transcription factor whose effects include either cell-cycle arrest, until damage has been repaired, or apoptosis if damage is too extensive for repair.
- Other effectors include cyclin-dependent kinases, protein kinases that regulate the cell cycle.
Connection Inaccurate replication past damaged sites in DNA leads to mutation, and this is the primary pathway by which UV irradiation causes skin cancers.
9.) Recombination
- Population genetics teaches us that the survival of a species depends on its ability to maintain genetic diversity, so that individuals can vary in their ability to respond to unforeseen environmental pressures.
- Diversity is maintained through mutation, which alters single genes or small groups of genes in an individual, and recombination, which redistributes the contents of a genome among various progeny during reproduction.
- As initially described by cytogeneticists, recombination results from crossing over between paired homologous chromosomes during meiosis in eukaryotes.
- Recombination, however, encompasses more processes and biological functions than those involved in sexual reproduction.
- Strictly speaking, recombination is any process that involves the formation of new DNA from distinct DNA molecules, such that genetic information from each parental DNA molecule is present in the new molecules.
- The double-strand break repair and daughter-strand gap repair processes described earlier are forms of recombination.
- So, too, is the formation of recombinant genotypes in bacterial conjugation—and, of course, recombinant DNA molecules constructed in vitro.
Concept Recombination is any process that creates end-to-end joining from two different DNA molecules.
- We begin our discussion with site-specific recombination, which was initially studied as a model for homologous recombination;
- It is important for the insight it provides into infection of cells by some viruses, including retroviruses.
10.) Site-Specific Recombination
- Many DNA viruses maintain a long-term relationship with the cells that they infect by inserting their genome into one or more chromosomes of the infected cell.
- Retroviruses carry out a similar process, which was outlined for HIV in Chapters 20 and 22; in this process, the RNA genome makes a DNA copy, which can then become inserted into host-cell chromosomes.
- The mechanism of viral genome integration is best understood for bacteriophage λ, and that is what we describe here.
- Phage T4 is a lytic phage.
- After infecting a bacterial cell, a cycle of virus multiplication always occurs, followed by lysis of the cell, which releases hundreds of new viral particles.
- Phage λ, by contrast, is a temperate phage, which does not always kill its host. Infection can lead either to a lytic growth cycle, like T4, or a lysogenic response, in which the viral chromosome becomes linearly inserted into the bacterial chromosome; once inserted, it can remain in a quiescent state, called a prophage, for many generations.
- A change in environmental conditions can trigger the expression of genes that direct excision of the viral chromosome from that of the host and turn on genes leading to virus multiplication and eventual lysis of the cell.
- Understanding these processes contributed importantly to our understanding of transcriptional regulation, which we will discuss in Chapter 26.
- The phage λ genome, as it exists in phage particles, is a linear duplex DNA molecule with short single-stranded tails at each 5′ end.
- These are called cohesive ends, or sticky ends, because they are complementary in sequence.
- Hence, the molecule can circularize by base pairing between the ends.
- The resultant nicks are closed by DNA ligase. In an infection leading to lysogeny, the covalently closed λ circle becomes linearly inserted into the E. coli host-cell chromosome, as schematized in Figure 23.15.
- A virally encoded protein called integrase recognizes specific sequences in the viral and bacterial chromosomes, which are brought together by their joint attraction for integrase.
- Unlike homologous recombination, which requires several hundred base pairs of sequence homology in order to occur, site-specific recombination requires fewer than two dozen homologous base pairs.
- In the case of phage λ (see Figure 23.15), sequences in the phage and bacterial genomes, called attP and attB, respectively, bind to specific sites on the integrase molecule, which catalyzes strand breaking and resealing reactions that linearly insert the viral chromosome into the chromosome of the infected E. coli cell.
- In this process, the sites of DNA recognition, breakage, and joining are brought about by specific DNA–protein interactions rather than by the DNA–DNA interactions via extensive sequence homology that categorize homologous recombination.
11.) Homologous Recombination
Breaking and Joining of Chromosomes
- The most straightforward way to accomplish recombination is to break and rejoin DNA molecules.
- However, if recombination occurs this way, the sites of breakage must be precisely the same on both recombining chromosomes for intact genes to be regenerated.
- Some researchers favored alternative mechanisms, but in 1961 Matthew Meselson and Jean Weigle showed that recombination in fact does occur via breakage and rejoining of chromosomes.
- The demonstration involved a Meselson–Stahl type of experiment (see Figure 4.17).
- E. coli was infected with two genetically distinct λ phage populations, one of which had been density labeled by growth in 13C–15N medium.
- The phage particles resulting from this mixed infection were centrifuged to equilibrium in a cesium chloride gradient.
- Phages with recombinant genotypes were recovered from all parts of the gradient, whereas nonrecombinant phages were uniformly light or heavy.
- This result means that recombinant phages contained DNA derived from both parents, which could have occurred only by breaking and rejoining DNA strands.
- Recombination is stimulated by processes that nick or break DNA strands, such as thymidine starvation or UV irradiation.
- This suggested that single-stranded DNA, or free DNA ends, play a role in initiating recombination.
Models for Recombination
- In 1964, Robin Holliday proposed a model for homologous recombination between duplex DNA molecules.
- Detailed in Figure 23.16, Holliday’s model continues to dominate thinking and experiments about this process.
- Holliday proposed that recombination begins with nicking at the same site on two paired chromosomes (step 1).
- Partial unwinding of the duplexes is followed by strand invasion, in which a free single-strand 3′ end from one duplex pairs with its unbroken complementary strand in the other duplex, and vice versa (step 2).
- Enzymatic ligation generates a crossed-strand intermediate, called a Holliday junction (step 3).
- The crossed-strand structure can move in either direction by duplex unwinding and rewinding (branch migration, step 4).
- The Holliday junction “resolves” itself into two unbroken duplexes by a process of strand breaking and rejoining.
- The process leading to recombination begins with isomerization of the Holliday structure (step 5), followed by strand breakage, so that the strands that break (in step 9) are those that were not broken in step 1.
- Resolution of the resulting structure (steps 10 and 11) generates two chromosomes recombinant for DNA flanking the region and each containing a heteroduplex region (each strand from a different parental molecule).
- However, if the original crossed strands (those that were broken in step 1) break and rejoin (steps 6, 7 and 8), the products are nonrecombinant duplexes, each containing a heteroduplex region (i.e., nonrecombinant with respect to the outside markers A and Z).
Concept Homologous recombination breaks and rejoins chromosomes.
- Considerable evidence now supports the central tenets of the Holliday model, including electron microscopic visualization of Holliday junctions (Figure 23.17).
- However, a major problem with the Holliday model was the requirement that two duplex DNAs undergo single-strand nicking at precisely the same points.
- How might that problem be resolved?
- Matthew Meselson and Charles Radding proposed that recombination could start with a single nick. The 3′ end of the nick invades the unbroken duplex and initiates strand displacement synthesis, with the displaced single strand then pairing with its partner in the unbroken strand, as indicated in Figure 23.18.
Proteins Involved in Homologous Recombination
- The Holliday and Meselson–Radding models explained most of the existing data on homologous recombination between paired chromosomes, particularly as studied in lower eukaryotes such as yeast.
- Moreover, the models could easily be adapted to explain daughter-strand gap repair or the recombination that occurs in bacteria after transformation or conjugation. Some of the proteins thought to participate, notably DNA polymerase, DNA ligase, and single-strand DNA-binding protein, had been characterized and shown to participate in recombination.
- What other proteins might participate in recombination if the models are largely correct?
- To answer this question, we turn back to E. coli and its phages and the characteristics of bacterial mutants defective in recombination.
- Mutations conferring a recombination-defective (rec−) phenotype map in several loci, and two important proteins are responsible for most bacterial recombination events.
- One of these gene products, the RecA protein, was introduced earlier.
- The other protein is called exonuclease V, or the RecBCD nuclease.
- RecA is a multifunctional protein with molecular weight of about 38,000.
- As we will see in Chapter 26, it participates in regulating the SOS response. In recombination, it promotes the ATP-dependent pairing of homologous strands, as mentioned earlier in connection with daughter-strand gap repair.
- As schematized in Figure 23.19 (left panel), the process begins with a reaction between RecA and single-stranded (ss) DNA, to give a RecA-ssDNA filament.
- RecA wraps about ssDNA as a multisubunit right-handed helix, with six RecA monomers per turn.
- Once ssDNA is bound, the filament searches double-stranded (ds) DNA, seeking sequences complementary to those in the single strand already bound.
- In this process, dsDNA is also taken up within the filament, giving a “joint molecule” (Figure 23.19, center panel).
- Binding to dsDNA requires ATP, but that ATP need not undergo hydrolysis.
- By contrast, movement of the ssDNA-protein complex with respect to the dsDNA does require ATP hydrolysis.
- During this process, dsDNA becomes underwound and stretched to about 1.5 times its normal length.
- The complex moves in a 5′-to-3′ direction along the initially bound ssDNA. During the movement, a triple-stranded structure transiently forms, with ssDNA (the red strand in Figure 23.19) wound in the minor groove of the dsDNA.
- The ssDNA continually tests the antiparallel strand in dsDNA (yellow) for sequence complementarity.
- It is not clear how this occurs, but evidently short oligonucleotide sequences swing out from the duplex structure and can pair with ssDNA if sequence complementarity is found.
- Once complementarity is established, branch migration occurs, with simultaneous strand exchange.
- A duplex is formed between the red strand (ssDNA originally) and the yellow strand (complementary to the red strand), while the displaced (green) strand is spooled out, away from the complex.
- ATP hydrolysis during this period may promote rotation of the DNA within the filament, facilitating the release of the displaced strand.
Concept RecA, a multifunctional bacterial enzyme, uses ATP to promote the pairing of homologous DNA sequences.
- Recombination occurs preferentially at or near particular DNA sequences.
- In E. coli, recombination is favored near a particular octanucleotide sequence, 5′-GCTGGTCC, called Chi (for crossover hotspot instigator).
- How does this site act to stimulate recombination?
- The RecBCD protein, a multifunctional heterotrimeric enzyme encoded by the recB, recC, and recD genes, displays sequence specificity for Chi.
- This enzyme binds at a double-strand break on duplex DNA and uses two helicase activities—RecB and RecD—to unwind and partially degrade the DNA.
- As shown from the crystal structure of a RecBCD-DNA complex, both helicases are in contact with DNA (Figure 23.20).
- The RecD helicase activity is higher than that of RecB, so as the protein moves, the 3′ end is displaced as a single-stranded loop ahead of RecB and becomes coated with SSB protein.
- Both strands are degraded by associated nucleases, but because of the differential speeds of the protein motors, more of the 3′ end is saved as the loop.
- When the enzyme reaches Chi, the protein pauses briefly, and a sequence-specific interaction causes RecBCD to switch speeds and change its preferred polarity of DNA degradation.
- As RecB moves faster, the 3′-terminated loop is reeled in by RecB and coated with RecA.
- An associated nuclease releases the bound 3′ end (not shown), freeing it for strand invasion of a neighboring duplex.
- Once a Holliday junction is formed, branch migration is essential for eventual formation of recombinant structures, as was shown in Figure 23.16.
- This is largely the responsibility of three other proteins.
- In E. coli, these three proteins are products of the ruvA, ruvB, and ruvC genes.
- RuvA is a DNA-binding protein, whose specificity directs it toward the four-stranded Holliday structure.
- RuvB protein is an ATP-requiring motor protein, which binds to two opposed arms of the junction.
- In the model shown in Figure 23.21, which is based on crystal structures of the isolated RuvA and RuvB proteins, the two RuvB molecules act as twin pumps, rotating the two arms in opposite directions.
- This forces branch migration by driving the rotational movement of the other two strands toward the junction.
- Eventually, RuvC binds and begins the resolution of the Holliday structure by nicking two strands.
Concept RecBCD, a multifunctional enzyme, unwinds and rewinds DNA, with one strand being unwound more rapidly and converted to a single-stranded 3′ end.
- Although Ruv protein homologs have not yet been detected in eukaryotic cells, much of the biochemistry of homologous recombination in eukaryotes is similar to what is described here.
- In particular, the RAD51 protein of both human cells and yeast has a strand-pairing activity similar to that of RecA, and the two proteins show extensive sequence homology.
- As noted earlier, an essential function of homologous recombination in eukaryotic cells is the repair of double-strand breaks, both during meiosis and as a result of DNA damage.
- A broken chromosome can use the sequence information in its homolog to reconstruct the original DNA sequence at the site of the break, as was shown in Figure 23.14.
- Understanding the biochemistry of recombination is of far more than academic interest.
- In recent years, scientists have learned how to direct the insertion of DNA into specific sites in mammalian genomes by use of homologous recombination.
- This targeted insertion allows the creation of a “knockout mouse,” in which any desired gene can be inactivated to allow investigation of the biological function of the deleted gene (see Tools of Biochemistry 23A).
- The recent development of CRISPR-Cas9 technology has made it even simpler to obliterate any desired gene function (also described in Tools of Biochemistry 23A).
Connection The ability to knock out the expression of any gene in a mouse by targeted homologous recombination has led to important insights into human diseases including cancer, obesity, heart disease, diabetes, arthritis, aging, and Parkinson’s disease.